Мышечная дистрофия, или миопатия Дюшенна , — тяжелая наследственная патология, которая постоянно прогрессирует. Замедлить мышечное разрушение практически невозможно.
Связано это с врожденными изменениями. Впервые о миопатии Дюшенна заговорили в середине XIX века. Обнаружил эту патологию французский невролог. В тот момент был известен один тип течения болезни, через некоторое время выделили еще несколько способов развития состояния.
Этот тип болезни сильно похож на миодистрофию Беккера , но в то же время отличается от него сложностью и внешними признаками.
Миодистрофия Дюшенна обнаруживает у 1 ребенка из 4000. Этот тип патологии относится к самым распространенным мышечным дистрофиям, относится к врожденным заболеваниям.
Одному из генов в структуре генома человека присвоили имя невролога, в честь которого и было названо отклонение. На мышечную дистрофию Дюшенна могут влиять разные факторы:
- кровосмешение;
- предрасположенность генетического характера, например, при наличии миопатии Дюшенна у кого-либо из родни;
- неправильный синтез мышечных волокон, ускоренное распространение и замещение жировой прослойкой, соединительными волокнами;
- наследственные формы синдрома Дюшенна, чаще всего переходящие от матери;
- мутация генома при формировании во время беременности;
- аномалии в хромосомных структурах неясного происхождения;
- сильные нарушения в развитии дистрофина;
- патологические изменения биохимии в крови.

Также миопатия Дюшенна формируется при заболеваниях соединительной ткани, не связанных напрямую с генетическими отклонениями.
Характеристика наследственной патологии
Генетическая природа заболевания была сразу же доказана после обнаружения синдрома в 1868 году. Эта патология почти идентична с миодистрофией Беккера, то есть, обладает теми же генетическими предпосылками для формирования.
Однако миодистрофия Беккера отличается иными симптомами. Для болезни характерны следующие особенности:
- диагностируется у мальчиков до 5 лет;
- прогрессирует стремительно;
- у девочек никогда не обнаруживается;
- атрофия мышц обладает ступенчатым развитием – сначала страдает тазовый пояс;
- затем вовлекаются мышцы ног;
- после этого миопатия Дюшенна поражает мышцы спины, плеч;
- завершается прогрессирующая мышечная дистрофия Дюшенна поражением рук;
- специфический признак нарушения – деформация позвоночника, чаще встречающаяся в форме кифоза или лордоза;
- миодистрофия Дюшенна почти всегда сопровождается повреждениями грудины и стоп, они становятся неправильной формы, сильно меняют тело человека;
- при патологии, в отличие от миодистрофии Беккера, появляется повреждение левого сердечного желудочка, аритмия и кардиопатия;
- примерно у 30% пациентов развивается олигофрения.

Мышечная дистрофия Дюшенна никогда не протекает в легкой степени , всегда имеет крайне неблагоприятный прогноз. Развивается быстро, возможность ходить пациент утрачивает уже к 12 годам. При мышечной дистрофии Дюшенна смерть наступает из-за инфекции бронхов или легких, после остановки сердца.
Симптомы нарушения
Первые признаки миопатии Дюшенна встречаются уже в возрасте 1,5 лет. В редких случаях их не удается заметить до 5 лет. Проявляются признаки заболевания Дюшенна сначала в легкой степени. Их комбинация зависит от общего состояния здоровья:
- у ребенка возникает сильная неустойчивость, наблюдается неловкость в движении, он часто падает и очень медлителен;
- миопатия Дюшенна сопровождается тем, что во время ходьбы ребенок вихляет, постоянно спотыкается, в результате чего малыш боится вставать на ноги, возникает выраженная двигательная пассивность;
- со временем при мышечной дистрофии Дюшенна становится видна «утиная» походка с выпяченной вперед грудью и отведенными назад лопатками;
- если ребенок сидит или лежит, то принять стоячее положение при мышечной дистрофии Дюшенна становится сложно;
- при попытках принять стоячее положение, малыш словно встает на лесенку, поднимается вверх задом;
- возникает гипертрофия мышц, они заполняются жировой тканью;
- также миодистрофия Дюшенна захватывает работу сердца, в результате чего развиваются патологии и недостаточность;
- мышечная дистрофия Дюшенна часто сопровождается и другим признаком – появляются нарушения в биоптатах скелета;
- постепенно изменяется положение крупных суставов, начинается деформация стоп;
- миопатия Дюшенна в 100% случаев приводит к полной инвалидности пациента, ему требуется кресло;
- в 15 лет при мышечной дистрофии Дюшенна наступает глубокая инвалидизация, опасная остановкой сердца и хронические или постоянно рецидивирующие нарушения в легких.

На фоне мышечной дистрофии Дюшенна у маленького пациента развивается острая депрессия, которую дети с трудом переносят. Нередко причиной смертности при миодистрофии Беккера и Дюшенна становится суицид.
Диагностика заболевания
Мышечная дистрофия Дюшенна крайне тяжело поддается диагностики. Для этого привлекают комплекс методов. Первое, что нужно пройти при подозрении на миопатию Дюшенна, — это ЭКГ. Для подтверждения диагноза необходимо, чтобы анализ показал нарушения стенки левого желудочка.

Следующий этап – это определение уровня дистрофина, который не меняется в сторону обычной дистрофии. Также необходимо сдать кровь на биохимический анализ. Если есть миодистрофия Беккера или болезнь, названная в честь французского невролога, отмечается высокий уровень КФК.
Дополнительно нужно пройти ЭМГ, генодиагностику, а также биопсию мышц. Именно последний анализ позволяет установить болезнь с достаточно высокой точностью. Электромиография не уступает в эффективности с точки зрения постановки диагноза «мышечная дистрофия Дюшенна».

Тактика лечения заболевания
Чтобы лечение мышечной дистрофии Дюшенна было эффективным, нужно четко следовать намеченному плану после постановки диагноза. Излечению болезнь никогда не поддается полностью, но можно значительно облегчить жизнь пациента. Современная медицина способна замедлить миопатию Дюшенна следующими методами:
- Тактика при обнаружении болезни до 5 лет. Радикального лечения миодистрофии Дюшенна не требуется. Нужна генетическая консультация и постоянна поддержка родителей больного ребенка.
- Лечение миопатии Дюшенна до 8 лет. В этом случае нужна поддержка мышечных тканей. Врачи назначают глюкокортикостероиды для замедления прогресса болезни: «Преднизолон» или «Дефлазакорт».
- Терапия от 8 до 20 лет. В этом случае значительно ослабляются мышцы, мышечная дистрофия Дюшенна приводит ребенка к инвалидному креслу.
- Терапия от 20 лет. В этом случае препараты частично перестают действовать, прогрессируют заболевания дыхательных путей.
Миопатия Дюшенна требует постоянного приема некоторых групп витаминов (B, E) , а также кальция, гормонов анаболиков, калия и некоторые виды аминокислот. Обязательно при мышечной дистрофии Дюшенна назначают инъекции АТФ, «Ретаболила», глютаминовой кислоты.

Важно! Поддержать здоровье при мышечной дистрофии Дюшенна можно и другими методами – ЛФК и электрофорезом.
ЛФК проходят небольшими курсами с обязательным участием терапевта. Также врачи рекомендуют делать массаж. Для электрофореза при миопатии Дюшеннанеобходимо использовать такие вещества, как липаза, хлорид кальция, «Прозерин».

В тяжелых случаях всё лечение проводят в домашних условиях, если есть медицинские возможности для организации сложной терапии специальными приборами.
Обязательное условие для лечения миопатии Дюшенна – постоянное наблюдение у кардиолога. Также необходимо составить грамотное меню. При заболевании нужно есть много овощей, приготовленных на пару, фруктов, растительных жиров и нежирного мяса. Запрещено употребление алкоголя, кофеина и крепкого чая.
Последствия и осложнения
В 100% случаев миопатия Дюшенна сопровождается тяжелыми последствиями для организма и сильно укорачивает жизнь. Пациент всегда умирает от осложнений заболевания – остановки сердца или легочной инфекции.
Если мышечную дистрофию Дюшенна удалось обнаружить в раннем возрасте, есть шанс, что человек доживет до 30 лет. Но только при условии адекватной терапии и комплексного подхода. Среди осложнений миопатии Дюшена нередко выделяют остеопороз, поражения позвоночника и суставов, а также патологии пищеварительной системы.

Мышечная дистрофия Дюшенна – тяжелое генетическое расстройство, терапия которого не способна оградить человека от одного исхода – смерти. В некоторых случаях пациентам удается прожить больше 20 лет после постановки диагноза. В других случаях младенцы умирают в течение первого года жизни.
В современной неврологии существует огромное количество заболеваний, характер возникновений которых не поддаются рациональному объяснению со стороны специалистов. К таким болезням можно отнести группу таких недугов, как мышечная дистрофия. Всего разновидностей данной болезни девять, но обо всем по порядку…
Мышечная дистрофия - это хроническое наследственное заболевание в результате развития которого происходит поражение мышечной системы человека. Пораженные мышцы перестают нормально функционировать, истончаются в размерах, а в месте их расположения в организме постепенно нарастает жировая прослойка.
Разновидности мышечной дистрофии
В современной неврологии данный недуг классифицируется на девять различных болезней. Классификация заболевания связана с:
- локализацией мышечных нарушений;
- характеристиками болезни;
- агрессивностью развития;
- возрастом.
Так, мышечная дистрофия бывает:
- Дюшенна;
- миотоническая (болезнь Штейнерта);
- Беккера;
- Эрба Рота;
- юношеская форма дистрофии Эрба-Рота;
- плече - лопаточная лицевая форма (Ландузи-Дежерина);
- алкогольная миопатия;
- дистальная форма;
- миодистрофия Эмери -Дрейфуса.
Дистрофия Дюшенна
Наиболее известная форма прогрессирующая дистрофия Дюшенна (псевдогипертрофическая дистрофия, мерозин - негативная врожденная дистрофия). Данная разновидность недуга проявляется в детском возрасте начиная с двух до пяти лет. В первую очередь от данного недуга страдают мышцы нижних конечностей, которые несмотря на малоподвижный образ жизни у маленьких пациентов, постепенно увеличиваются в размерах. Данная особенность связана с нарастанием жировой ткани вместо мышц.

Слева здоровый ребенок, справа больной
Постепенно, по мере прогрессирования болезни она переходит в верхнюю часть и поражает мышцы верхних конечностей. Как правило, уже к двенадцатилетнему возрасту маленький пациент перестает полностью двигаться. Летальность у данного заболевания очень высока примерно 85–90% больных не доживают до 20-летнего возраста.
В группе риска находятся представители мужского пола, так как девочек данный недуг практически не затрагивает.
Болезнь Штейнерта
Врожденная дистрофия Штейнера не зря имеет название миотоническая, так как в результате прогрессирования болезни происходит слишком медленное расслабление мышц после их сокращения (такое явление называется миотонией).
Данное заболевание, в отличие от предыдущего распространено у взрослых, в возрасте от 20 до 40 лет. Имеют место быть и случаи прогрессирования болезни у детей, как правило, в младенческом возрасте, однако, это скорее исключение, чем правило.

На лицо признаки миотонии (приоткрытый рот и веки)
Гендерной зависимости болезнь не имеет и в равной степени от нее страдают как мужчины, так и женщины. Отмечается, что при недуге проявляется слабость мимических мышц лица, а также конечностей. Прогрессирование, в отличие от болезни Дюшенна происходит медленно.
Отличительная черта заболевания, возможность поражения не только мышц конечностей, но и внутренних мышц (сердечная мышца), что в свою очередь, представляет большую опасностью для жизни человека.
Болезнь Беккера
Данная форма болезни также долго прогрессирует, и пациент длительное время чувствует себя хорошо. Обострение недуга может произойти на фоне полученных травм или различных заболеваний нервной системы, которые своим течением ускорят развитие недуга.
В группе риска находятся люди низкого роста.
Болезнь Эрба-Рота и юношеская ее форма
Данное аутосомно - рецессивное заболевание развивается у пациентов после 20 лет, а юношеская форма у детей и подростков 11-13-летнего возраста. Прогрессирование болезни происходит по восходящему варианту, то есть сначала поражаются мышцы нижних конечностей, и постепенно происходит восхождение недуга к верхним конечностям.
Отличительной чертой недуга является наличие выступающих лопаток, которые по мере прогрессирования выступают более явно и очевидно.
Во время ходьбы отмечается переваливание пациента, выпячивание живота и задвигание грудной клетки.
Болезнь Ландузи-Дежерина
Плече-лопаточно-лицевая форма данного заболевания является наиболее безразборным видом, так как поражает людей в возрасте от пяти до 55 лет. Для данного недуга характерно очень длительное развитие, пациент может сохранять трудоспособность до 25 лет жизни с данной болезнью.
Отличительными симптомами является поражение лицевых мышц, в результате чего у больного могут возникнуть проблемы с четкостью произношения, в связи с неполным смыканием губ. Кроме того, отмечается неполное смыкание век.
По мере развития у больного атрофируются лицевые мышцы, мышцы плеч, конечности и туловища.
Данная форма не зависит от генетической мутации.
Алкогольная миопатия
Данный вид болезни также несвязан с генными мутациями и причина его возникновения одна - злоупотребление алкоголем. У больных могут отмечаться болевые синдромы в конечностях, связанные именно с поражением мышц.
Различают острую и хроническую алкогольную миопатию.
Дистальная форма
Дистальная форма мышечной дистрофии является доброкачественным вариантом прогрессирующей дистрофии. Как правило, данное заболевание трудно дифференцировать от невральной амиатрофии Мари-Шарко. Для разделения этих двух заболеваний требуется проведение электроэнцефалограмму головы, которая и дает понимание с какой болезнью предстоит иметь дело.
К основным симптомам недуга относят атрофию мышц конечностей с их последующим исхуданием. Возможны парезы стоп, кистей рук и т. п.
Миодистрофия Эмери -Дрейфуса
Данный тип болезни первоначально, вообще, не был выделен в качестве отдельного заболевания, так как по своей симптоматике был схож с дистрофией Дюшенна. Однако, в дальнейшем в результате длительного исследования было установлено, что болезнь Эмери -Дрейфуса обладает индивидуальной симптоматикой.
Болезнь относят в разряд редких. В группе риска находятся люди до 30 лет, как правило, юный возраст. Есть свидетельства о проявлении симптомов и после 30 лет, но они единичны.
Основное отличие данного вида болезни - это проблемы с мышцами сердца, которые в итоге могут стать причиной смерти. Кардиомиопатия при данном недуге не единственное отличие. Кроме проблем с сердцем, у пациентов наблюдают стандартные для дистрофии Дюшенна признаки, но в более щадящем режиме развития.
Причины
Негативная составляющая болезней нервной системы заключается в том, что они тяжело поддаются изучению. По этой причине не до конца известно о факторах, провоцирующих развитие той или иной формы мышечной дистрофии.
Основная причина, для формирования большинства подвидов данной болезни является генная мутация, в частности гена, который отвечает за синтез белка.
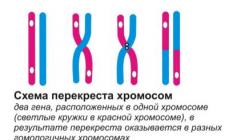
К примеру, болезнь Дюшенна имеет прямое отношение к мутации половой Х хромосомы. Основной переносчик нехорошего гена выступают девушки, которые несмотря на его наличие в собственной ДНК не страдают от данной болезни.
Что касается миотонической формы, то виновник ее возникновения ген, расположенный в 19 хромосоме.
Основные симптомы
Наличие большого количество различных подвидов у данного недуга указывает на различия в симптоматике, однако у болезни есть общие симптомы, которые в себя включают:

Диагностика мышечной дистрофии
Диагностические мероприятия при подобном недуге обширны, так как существует большое количество болезней, от которых необходимо дифференцировать болезнь.
На первоначальном этапе доктор обязательно изучит анамнез больного, уточнит о симптоматике, режиме дня и т. п. Эти данные требуются для составления плана проведения последующих диагностических мер, которые могут в себя включать:

Стоит отметить, что чем позже проявилась болезнь, тем лучше для больного, так как ранние симптомы в большинстве случаев заканчиваются смертью.
Лечение
Лечение мышечной дистрофии процесс сложный и затяжной, однако, в настоящее время не создано лекарство, которое полностью исцеляет больного. Все мероприятия направлены на облегчение жизни больного и восстановления некоторых утраченных способностей.
Для затормаживания развития болезни назначают следующие препараты:
- кортикостероиды;
- витамины В1;
- аденозинтрифосфат (АТФ).
Кроме того, для замедления процесса развития используют фетальные стволовые клетки, которые замедляют процесс дистрофии.
Помимо того, в качестве профилактических мероприятий назначают:
- массаж;
- физиотерапию;
- дыхательную гимнастику.
Помимо стандартных вариантов лечения, важно постоянно руководствоваться тремя главными составляющими в процессе жизнедеятельности:
- Адекватная физическая активность.
- Своевременная психологическая поддержка.
- Соблюдение диеты.
Физическая активность
Отсутствие желания у человека бороться с недугом оказывает негативное воздействие на организм. Судите сами, пассивность, нежелание двигаться угнетает и без того пораженную мышечную систему. Мышцам необходимо давать нагрузку, так как без нагрузки дистрофичные процессы начинают происходить быстрее, тем самым прогрессируя в более быстром темпе.
Умеренная физическая активность, использование поддерживающих приспособлений будут отличным подспорьем в борьбе с недугом.
При наличии болей в мышцах отлично подойдет плавание, йога, упражнения на растяжку.
Психологическая поддержка
Психологическая поддержка окружения важна для больного человека. А если недуг такой серьезный, как этот тем более. Для кого-то будет достаточно обычной поддержки со стороны друзей и родных, а кому-то может потребоваться квалифицированная психологическая помощь.
Важно дать понять такому человеку, что он не остался один на один со своей проблемой. Он должен понимать, что ему есть к кому обратиться, есть люди, которые сопереживают и поддерживают его.
Диета
Что касается режима питания и соблюдения диеты, существует расхожее мнение, что соблюдение противовоспалительной диеты может замедлить прогрессирование недуга. Данная диета снижает воспаление, уровень глюкозы, выводит токсины из организма и питает его полезными веществами.
Суть такой диеты в следующем:
- Отказ от продуктов, содержащих «плохие» жиры и замена их на «хорошие», введение в рацион ненасыщенных жиров, которые содержатся в оливковом, льняном, кунжутном масле, авокадо.
- Применение в пищу мяса и рыбы, при производстве которых не были использованы антибиотики или гормоны.
- Полное удаление из рациона рафинированного сахара и глютена.
- Употребление в пищу следующих продуктов - китайская капуста, брокколи, сельдерей, ананас, лосось, свекла, кукумария, имбирь, куркума и других продуктов, обладающих противовоспалительными свойствами.
- Молочные продукты допустимы только на основе овечьего и козьего молока.
- Допустимо употребление травяных чаев, лимонада, кваса, морсов и натуральных соков.
Профилактика
Так как мышечную дистрофию довольно тяжело предсказать и обнаружить на раннем этапе, профилактические мероприятия сводятся к двум простым рекомендациям:
Обязательное обследование женщины на этапе планирования беременности на предмет наличия мутаций в генах
В случае если до беременности, по каким-либо причинам не было сделано генетического обследования, уже в период беременности проводится тест на определение у плода мутаций в Х хромосоме.
Прогноз и осложнения
В зависимости от вида болезни прогноз может быть разным, и тем не менее можно выделить несколько возможных осложнений, возникающих при данном недуге.
- нарушения сердечной деятельности
- искривление позвоночника
- снижение интеллектуальных способностей больного
- снижение двигательной активности
- нарушения дыхательной системы
- летальный итог
Итак, мышечная дистрофия довольно опасное и неизлечимое заболевание, поэтому будущим родителям необходимо с глубокой ответственностью относиться к планированию беременности. Берегите себя и своих будущих детей!
Мышечная дистрофия Дюшенна
Мышечная дистрофия Дюшенна
(МДД
) - это серьезное рецессивное заболевание, которое характеризуется быстрым прогрессированием мышечной дистрофии
, которая в конечном итоге приводит к полной потере способности двигаться и смерти больного.
Это заболевание поражает примерно 1 человека из 4000, то есть - это наиболее распространенный тип мышечной дистрофии. Обычно на МДД болеют только мужчины, хотя женщины могут иногда быть носителями заболевания. Если же отец болен МДД, а мать является носителем, или тоже больна, то в таком случае на мышечную дистрофию Дюшенна может заболеть женщина. Расстройство возникает в связи с в дистрофин , который у людей расположен на (Xp21). Ген дистрофин кодирует деятельность белка дистрофина, который является важной структурной составляющей мышечной ткани. Дистрофин обеспечивает структурную устойчивость дистрофин-ассоциированного-гликопротеинового комплекса (ДАГ комплекса), расположенного на клеточной мембране.
Симптомы заболевания обычно появляются у детей мужского пола до 5 лет и могут проявиться в раннем детстве. Первыми признаками болезни являются прогрессирующая проксимальная слабость мышц ног и таза, связанная с потерей мышечной массы. Постепенно эта слабость распространяется на руки, шею и другие части тела. Ранние признаки расстройства могут также включать псевдогипертрофию (увеличение икроножных мышц и дельтовидных мышц), низкую выносливость и трудности при стоянии без посторонней помощи, как правило, человек также не может самостоятельно подняться. В процессе прогрессирования заболевания мышечная ткань постепенно заменяется жировой и фиброзной тканями (как следствие развивается фиброз). Для помощи при ходьбе, в возрасте 10 лет может быть необходимым применение специальных подтяжек, но большинство пациентов уже старше 12 лет не могут передвигаться без инвалидной коляски.
Позже возникают следующие признаки расстройства: аномалии развития костей, которые приводят к деформации скелета, в том числе к искривлению позвоночника.
В связи с прогрессивным ухудшением работы мышц, особь теряет возможность двигаться, что в конечном счете, может быть причиной паралича. Относительно отклонений в психическом развитии, то их наличие зависит от каждого конкретного случая, но если определенные отклонения и присутствуют, то они несущественно влияют на развитие ребенка, ведь со временем нарушение не прогрессирует. Средняя продолжительность жизни больных МДД варьирует от подросткового возраста до 20 - 30 лет. Известны случаи, когда больные доживали до 40 лет, но, к сожалению, такие случаи являются скорее исключением.
Распространенность
Мышечная дистрофия Дюшенна возникает в связи с мутациями в гене дистрофин, который расположен на Х-хромосоме. В связи с этим, ДМД встречается у 1 человека на 4000 новорожденных мужского пола. Мутации в гене дистрофин могут быть или возникают спонтанно во время зародышевой линии передачи.
Эпоним
Заболевание названо в честь французского невропатолога Жульема Бенджамина Аманда Дюшенна (Guillaume Benjamin Amand Duchenne), который впервые описал это заболевание в 1861 г оду.
Патогенез
Мышечная дистрофия Дюшенна обусловлена мутацией в гене дистрофин , которого Xp21. Дистрофин отвечает за соединение цитоскелета каждого мышечного волокна с основной базальной пластинкой (внеклеточного матрикса) через белковый комплекс, который состоит из многих субъединиц. Отсутствие дистрофина приводит к проникновению избыточного кальция в сарколему (клеточную мембрану). Как следствие изменения этих сигнальных путей, вода наполняет митохондрии, которые после этого разрываются. При дистрофии скелетных мышц, митохондриальная дисфункция приводит к усилению стресса вызванного цитозольным-кальциевым сигналом и усилению производства стресс-индуцированных активных форм кислорода (АФК). В этом сложном каскадном комплексе, который включает в себя несколько реакций еще до сих пор не понятно до конца, почему из-за повреждения сарколеммы увеличиваются проявления окислительного стресса, который в итоге приводит к смерти клетки. Мышечные волокна подвергаются некрозу и, наконец, происходит замена мышечной ткани жировой, а также соединительной.
Симптомы
Основным симптомом мышечной дистрофии Дюшенн
а - является мышечная слабость
, которая в первую очередь связана с атрофией мышц, а именно скелетной мышечной ткани. В первую очередь атрофируются мышцы бедер, таза, плеч и икроножные мышцы. Мышечная слабость возникает также в руках, шее и других частях тела, но обычно не так рано, как в нижней части тела. Икры часто увеличены. Обычно симптомы появляются в возрасте до 6 лет, но могут впервые проявиться еще в раннем детстве.
Другие физические симптомы
расстройства:
- неуклюжая (тяжелая) походка, шаги или бег (как правило, пациенты, ходят на пальцах (на носочках), через повышенный тонус икроножных мышц). Кроме того, такая манера ходить, является своеобразной адаптацией к постепенной потере функций колен;
- больные часто падают;
- постоянная усталость;
- у больного возникают трудности при осуществлении таких двигательных навыков как бег или скачки;
- усиление поясничного лордоза, которое приводит к атрофии (уменьшение размеров) мышц сгибателей бедра. Она влияет, в общем, как на осанку, так и на манеру ходить и бегать, в частности;
- мышечные контрактуры, которые существенно уменьшают функциональность ахилового и подколенного сухожилия, поскольку количество мышечных волокон уменьшаются и возникает фиброз мышц;
- прогрессирование трудности при ходьбе;
- деформация мышечных волокон;
- дсевдогипертрофия (увеличение) языка и икроножных мышц, вызванная заменой мышечной ткани жировой и соединительной;
- повышенный риск нейроповеденческого расстройства (такого как синдром дефицита внимания и гиперактивности (СДВГ), расстройства аутистического спектра), трудности с учебой (дислексия) и не прогрессирующие отклонения в определенных когнитивных функциях (в частности, таких как краткосрочная словесная память), которые, как считают ученые, возникают из-за отсутствия или нарушения функционирования дистрофина в мозге;
- возможная потеря способности ходить (обычно в возрасте до 12 лет);
- скелетные деформации (в некоторых случаях возникает сколиоз);
Признаки и тестирования
Как уже было сказано, атрофия мышц при МДД начинается в виде мышечной слабости в ногах и тазовом поясе, затем переходит к мышцам плеч и шеи, после чего, повреждает мышцы рук и дыхательные мышцы. Важным видимым признаком в начале развития заболевания является увеличение икроножных мышц (псевдогипертрофии ). Распространенным явлением есть кардиомиопатия, но развитие сердечной недостаточности или аритмии (заболевания связаны с нарушениями ритма сердца, последовательности и силы сокращений сердечной мышцы) встречаются довольно редко.
Наличие симптома Говерса отражает более тяжелые нарушения мышц нижних конечностей. Можно говорить о наличии симптомов, в случае, если ребенок помогает себе встать с помощью рук: сначала, ребенок становится на четвереньки (опираясь на пол ногами и руками), а затем, держась руками за ноги, контролирует направление своего движения;
- больные МДД дети, часто устают быстрее и имеют меньше силы, чем их сверстники;
- очень высокий уровень креатин-киназы (КФК-ММ) в крови тоже может стать показателем развития и прогрессирования заболевания;
- при проведении электромиографии (ЭМГ) видно, что слабость организма вызвана повреждением мышечной ткани, а не повреждением нервной проводимости;
- может выявить генетические нарушения в гене Xp21;
- мышечная биопсия с последующим гистологическим, иммуногистохимическим или имуноблотинговим исследованием) или генетическое тестирование (с помощью анализа крови), подтверждает отсутствие дистрофин.
Диагностика
ДНК-тест
Мышечно-специфическая изоформа гена дистрофина состоит из 79 экзонов. Тестирование и их анализ, как правило, позволяют определить тип мутации экзона или определить какие экзоны повреждены. Анализ ДНК в большинстве случаев подтверждает предварительную диагностику другими методами.
Биопсия мышц
Если при анализе ДНК никаких мутаций не обнаруживается, то возможно проведение мышечной биопсии. Для этой процедуры с помощью специального инструмента берут маленький образец мышечной ткани и, использовав специальный краситель, определяют наличие /отсутствие в мышечной ткани дистрофина. Полное отсутствие белка указывает на наличие этого заболевания.
За последние несколько лет ДНК-тесты были существенно усовершенствованы, на сегодня они проявляют больше мутаций и поэтому мышечную биопсию для подтверждения МДД сейчас используют все реже.
Пренатальное тестирование
Если один или оба родителя являются "носителями" этого заболевания, то существует риск того, что их еще не народившийся ребенок будет поражен этим расстройством. Для определения того, будет будущий ребенок больным МДД, используют методы . На сегодня эти методы доступны только для определения некоторых нервно-мышечных расстройств. Различные пренатальные тесты могут проводиться примерно на 11 недели беременности.
Исследование с помощью биопсии хориона
(CVS) можно проводить на 11-14 неделях, амниоцентез
можно использовать после 15 недели, забор крови плода
возможен примерно на 18 неделе. Родители должны внимательно изучить все возможные методы и, возможно, с помощью выбрать наиболее оптимальный для себя вариант. Если тестирование будет осуществлено на ранних сроках беременности, то это позволит досрочно прекратить беременность, в случае наличия заболевания у плода, однако, при применении таких методов, увеличивается риск выкидыша при последующих беременностях, чем при тех методах, которые применяются позже (около 2% , по сравнению с 0,5%).
Лечение
Никаких известных эффективных препаратов для лечения мышечной дистрофии Дюшенна, не существует. Хотя согласно последним исследованиям стволовых клеток существуют перспективные векторы, которые могут заменить поврежденные мышечные ткани. Однако, на данном этапе лечения, как правило, симптоматическое и направлено на улучшение качества жизни больного человека.
Оно включает в себя:
Употребление таких кортикостероидов как преднизолон и дефлазакорт для увеличения энергии и сил и облегчения тяжести некоторых симптомов;
- рандомизированные контролируемые исследования показывают, что использование бета 2-агонистов увеличивает мышечную силу, но не замедляет процесс прогрессирования заболевания. Время контроля за людьми, которые употребляли бета 2-агонисты составляет около 12 месяцев, следовательно, результаты этих испытаний не могут быть экстраполированы на больший период времени;
- рекомендуется умеренная физическая активность, разрешается заниматься плаванием. Бездействие (например, постельный режим) может усилить прогрессирование заболевания;
- для поддержания мышечной силы, гибкости и функциональности суставов важна физиотерапия;
- использование ортопедических приспособлений (например, инвалидных колясок) может улучшить способность больного двигаться и самостоятельно обеспечивать свои потребности. Использование так называемых съемных стяжек, фиксирующих голень во время сна позволяет отложить начало контрактур (ограничение движений суставов).
- по мере прогрессирования заболевания необходимым становится использование специальных респираторных механизмов, позволяющих обеспечить нормальный процесс дыхания.
Центром по контролю и профилактике заболеваний (Centers for Disease Control and Prevention (CDC)) были разработаны общие многопрофильные стандарты (принципы) помощи больным МДД. Эти принципы были опубликованы в двух частях в журнале The Lancet Neurology в 2010 году. (Http://www.treat-nmd.eu/patients/DMD/dmd-care).
Прогноз
Мышечная дистрофия Дюшенна повреждает все скелетные мышцы, мышцы сердца и дыхательные мышцы (на более поздних стадиях). Больные МДД, как правило, живут только к подростковому возрасту или умирают в возрасте 30-40 лет. Последние достижения в области медицины, позволяют надеяться на увеличение продолжительности жизни больных этим расстройством.
Иногда (но очень редко) особи с МДД доживали до 40-50 лет, но лишь с помощью использования надлежащего дополнительного оборудования (инвалидных колясок и кроваток), вентиляционной поддержки дыхания (с помощью трахеостомии или специальной дыхательной трубки), очистки дыхательных путей и принятие необходимых сердечных препаратов. Кроме того, для увеличения продолжительности жизни необходимо на ранних этапах заболевания спланировать механизм ухода за больным на более поздних этапах.
Физиотерапия
Физиотерапия при МДД в основном направлена на больных детей и на развитие их максимального физического потенциала. Цель физиотерапии состоит в следующем:
Минимизировать развитие контрактур и деформаций путем разработки соответствующей программы для сохранения эластичности мышц, а также возможна разработка программы физических упражнений;
Предусмотреть и минимизировать появление вторичных физических осложнений;
Контролировать дыхательные функции и предоставление информации относительно того, какие техники и методы дыхательных упражнений необходимо применять, чтобы очистить дыхательные пути от выделений;
Механическая вентиляция (респираторная помощь)
Использование современных аппаратов искусственной вентиляции легких, которые доставляют регулируемый объем (количество) воздуха в легкие человека, особенно важно это для людей, страдающих от дыхательных проблем, возникающих в процессе развития мышечной дистрофии. Применение этих механизмов при заболевании МДД можно начать в подростковом возрасте, когда дыхательные мышцы начинают повреждаться. Однако, известны случаи, когда даже в возрасте 20 лет, больные не нуждались в использовании таких аппаратов.
Если жизненная емкость легких упала ниже 40% от нормы, то эти респираторы могут использоваться во время сна, ведь именно в это время заболевший может больше всего пострадать от гиповентиляции .
Гиповентиляция во время сна определяется тщательным исследованием истории этого расстройства сна, путем проведения оксиметрии и измерением количества углекислого газа в капиллярной крови. Для вентиляции необходимым может быть проведение процедуры интубации или трахеотомии трубки, через которые воздух непосредственно доставляется в легкие, однако, для некоторых людей вполне достаточно того воздуха, который поступает при использовании специальной маски.
Если жизненная емкость легких продолжает снижаться и составляет менее 30% от нормы, то необходимым становится увеличение продолжительности использования аппарата искусственной вентиляции легких (эта продолжительность должна увеличиваться, по мере необходимости). Трахеотомическая трубка может использоваться как в дневное время, так и во время сна, однако возможна также ситуация, когда достаточным является и количество воздуха, поступающего через дыхательную маску. Аппарат искусственной вентиляции легких легко помещается на вентиляторном лотке снизу или сзади инвалидной коляски, для большей портативности этот механизм можно обеспечить специальной энергетической батареей.
Проводимые исследования
Для выявления лекарств, которые бы позволили смягчить последствия действия МДД или же вообще вылечить ее сегодня ведутся весьма перспективные исследования. Существует много направлений этих исследований, особенно стоит отметить лечения стволовыми клетками, технологию пропуска-экзонов, аналоговую активацию и генную замену. Другим направлением исследований является поддерживающая терапия, которая направлена на разработку лекарств, которая бы предотвратить развитие и прогрессирование болезни.
Лечение стволовыми клетками
Ученые считают, что стволовые клетки, выделенные из мышц (клетки-сателлиты) имеют способность превращаться в миоциты. При непосредственном введении в мышцы животных, они не могут распространяться по всему организму. И для того, чтобы такая терапия была эффективной, необходимо осуществлять инъекции в каждую мышцу через каждые 2 мм. Этот недостаток лечебной процедуры можно исправить, использовав другие, мультипотентные стволовые клетки, которые называются перициты. Они расположены в кровеносных сосудах скелетных мышц.
Эти клетки можно ввести в организм системно и усваиваются они организмом путем попадания в кровоток. Попав, в сосудистую сетку перициты сливаются, образуя миотубулы. Это означает, что они могут быть введены артериально, далее они попадают через стенки сосудов в мышцы. Эти данные свидетельствуют о наличии потенциала для проведения клеточной терапии МДД. Небольшое количество перицитов может быть получена из организма человека, затем они могут быть выращены искусственным путем и введены в кровоток после чего, как считают исследователи, существует вероятность того, что они могут найти «свой путь» к пораженным мышцам.
Активация утрофина
Регулирования экспрессии утрофина для лечения МДД представляет большой интерес, поскольку именно этот является ближайшим эндогенным аналогом дистрофина в . Этот ген короче и расположен у человека на . Исследователи в настоящее время сфокусированы на понимании регулирования за свое выражение в клетках. Еще ранее стало известно, что активация утрофину может частично компенсировать недостаток дистрофин в мышечных клетках. Последние лабораторные исследования утрофину показали существенное улучшение роста мышц у мышей с ДМД. Дальнейшие исследования, которые будут проводиться с участием людей могут дать ответ на вопрос относительно того, действительно активация утрофину у людей больных МДД повысит качество и продолжительность их жизни.
Нуклеотиды были использованы для коррекции нарушений сплайсинга в клетках, полученных от лиц, больных бета-талассемией и использованы для исследования их действия при лечении МДД, спинальной мышечной атрофии, синдрома Хатчинсона-Гилфорда и других заболеваний.
Для лечения лиц больных МДД, использование AONs, согласно исследованиям может быть очень перспективным. Например, МДД может возникать в результате изменений мРНК, вызванных сдвигом рамки считывания (например, вставки или сплайс-мутаций). Предполагается, что в случае если заболевание вызвано, этими нарушениями, то оно может быть излечено путем восстановления последовательности мРНК, то есть возвращение рамки считывания на нужное место. Для того чтобы это сделать необходимо, чтобы AON-и помогли выявить определенные регионы пре-мРНК, которые бы помогли замаскировать распознавания Сплайсосома экзона или экзонов.
И хотя использование AON-ов - может быть достаточно перспективным, но одной из основных проблем является их постоянное возвращение в мышцы. Методы постоянного системного поступления на сегодня испытываются на людях.
Кроме того, исследуются также новые методы, которые бы позволили обойти все недостатки выше описанной процедуры. Эта терапия состоит в изменении U7 малой ядерной РНК в 5 положении еще до в целевых регионах пре-мРНК. Этот метод действует на мышах, пораженных МДД.
Этиология и встречаемость мышечной дистрофии Дюшенна . Мышечная дистрофия Дюшенна (MIM №310200) - панэтническая Х-сцепленная прогрессирующая миопатия, вызванная мутациями в гене DMD. Встречаемость составляет приблизительно 1 на 3500 новорожденных мальчиков.
Патогенез мышечной дистрофии Дюшенна . Ген DMD кодирует дистрофии, внутриклеточный белок, экспрессирующийся преимущественно в гладких, скелетных и сердечной мышце, а также в некоторых нейронах мозга. В скелетной мускулатуре дистрофии составляет часть большого комплекса связанных с сарколеммой белков, обеспечивающих устойчивость сарколеммы.
Мутации в гене DMD , вызывающие миодистрофию Дюшенна, включают крупные делеции (60-65%), крупные дупликации (5-10%) и небольшие делеции, инсерции или замены нуклеотидов (25-30%). Самые крупные делеции происходят в одной из двух горячих точек. Нуклеотидные замены встречаются по всему гену, преимущественно в динуклеотидах CpG.
De novo мутации возникают со сравнимой частотой в ходе овогенеза и сперматогенеза; наиболее крупные делеции de novo возникают в овогенезе, тогда как большинство de novo нуклеотидных замен возникает в ходе сперматогенеза.
Мутации , вызывающие фенотипическое отсутствие дистрофина, приводят к более тяжелому поражению мышц, чем мутантные аллели DMD, экспрессирующие частично функциональный дистрофии. Корреляции между генотипом и фенотипом для интеллектуального снижения не обнаружено.
Фенотип и развитие мышечной дистрофии Дюшенна
Мужчины с мышечной дистрофией Дюшенна . Миодистрофия - прогрессирующая миопатия, приводящая к дегенерации и слабости мышц. Начинаясь с мышц тазобедренного пояса и сгибателей шеи, мышечная слабость прогрессивно захватывает плечевой пояс и дистальные мышцы конечностей и туловища. Хотя изредка и выявляют случайно больных в период новорожденности за счет гипотонии или задержки развития, обычно больных мальчиков диагностируют в возрасте от 3 до 5 лет при появлении аномалий походки.
К 5 годам большинство пораженных детей используют приемы Говерса и имеют псевдогипертрофию мышц голеней, т.е. увеличение голеней вследствие замены мышц жировой и соединительной тканью. К возрасту 12 лет основная часть больных обездвижены в инвалидном кресле и имеют контрактуры и сколиоз. Большинство пациентов умирают от нарушения легочной функции и пневмонии; средний возраст смерти - 18 лет.
Почти 95% больных миодистрофией Дюшенна имеют те или иные кардиологические отклонения (дилатационная кардиомиопатия или электрокардиографические аномалии), а 84% имеют видимые поражения мышцы сердца при вскрытии. Хронические нарушения сердца бывают почти у 50% пациентов, изредка сердечная недостаточность вызывает у них жалобы. Хотя дистрофии также присутствует в гладких мышцах, гладкомышечные осложнения встречаются редко и включают расширение желудка, заворот кишок и гипотонию мочевого пузыря.
Больные миодистрофией Дюшенна имеют IQ примерно на 1 среднеквадратичное отклонение ниже обычного, и почти треть имеет ту или иную степень умственной отсталости. Причины этого не установлены.
Женщины с мышечной дистрофией Дюшенна
Возраст начала и тяжесть миодистрофии Дюшенна у женщин зависят от степени смещения инактивации Х-хромосомы. Если Х-хромосома, несущая мутантный аллель DMD, активна в большинстве клеток, у женщины развиваются признаки миодистрофии Дюшенна; если преимущественно активна Х-хромосома, несущая нормальный аллель DMD, женщины имеют только несколько или не имеют вообще симптомов данного заболевания.
Независимо от того, есть ли у них клинические симптомы скелетной мышечной слабости , женщины-носительницы имеют отклонения в функции сердечной мышцы, например дилатационную кардиомиопатию, дилатацию левого желудочка и электрокардиографические изменения.
Особенности фенотипических проявлений дистрофии Дюшенна
:
Возраст начала: детство
Слабость мышц
Гипертрофия голеней
Небольшая интеллектуальная недостаточность
Высокий уровень креатинкиназы сыворотки
Лечение мышечной дистрофии Дюшенна
Диагноз мышечной дистрофии Дюшенна основывается на семейном анамнезе и ДНК-анализе или мышечной биопсии с иммуногистохимическим определением дистрофина.
В настоящее время излечение мышечной дистрофии Дюшенна невозможно, хотя улучшившееся симптоматическое лечение повысило средний срок жизни от конца детства до ранней зрелости. Цели терапии - замедлить развитие болезни, обеспечить мобильность, предотвратить или исправить контрактуры и сколиоз, контролировать массу тела и улучшить функции легких и сердца.
Глюкокортикоидная терапия может замедлять развитие заболевания в течение нескольких лет. Исследуют несколько видов экспериментального лечения, включая генную передачу. Большинство больных нуждается также в расширенном консультировании, так как имеют дело с психологическими эффектами хронической фатальной болезни.
Риск наследования мышечной дистрофии Дюшенна
Третья часть матерей, родивших единственного больного сына , сами являются носительницами мутаций в гене DMD. Тем не менее определение носительства остается трудной задачей, поскольку в настоящее время доступные молекулярные методы не обнаруживают небольшие мутации типа однонуклеотидных замен. Определение риска носительства в семьях без найденной делеции или дупликации основывается на анализе сцепления, серии анализов уровня креатинкиказы сыворотки крови и мозаичной экспрессии дистрофина в образцах биопсии мышц (из-за случайной инактивации Х-хромосомы). При консультировании в связи с оценкой риска повторения нужно принимать во внимание высокую частоту мозаицизма в половых клетках (приблизительно 14%).
Если мать - носитель, каждый сын имеет 50% риск развития мышечной дистрофии Дюшенна, а каждая дочь - 50% риск унаследовать мутацию DMD. Отражая случайную природу инактивации Х-хромосомы, дочери, унаследовавшие мутацию в гене DMD, имеют низкий риск мышечной дистрофии Дюшенна; тем не менее по причинам, полностью непонятным, риск сердечных аномалий может достигать 50-60%. Если мать не носительница по результатам ДНК-тестирования, у нее остается приблизительно 7% риска родить мальчика с мышечной дистрофией Дюшенна вследствие полового мозаицизма. Для этих матерей показаны генетическое консультирование и, возможно, пренатальная диагностика.
Пример мышечной дистрофии Дюшенна . А.И., 7-летний мальчик, проходит обследование в связи с легкой задержкой развития. У него затруднения при подъеме по ступенькам, беге, снижение силы и выносливости при интенсивной физической нагрузке. Его родители, два брата и сестра полностью здоровы; другие члены семьи аналогичных жалоб не имеют. При осмотре выявлены затруднения при подпрыгивании, приемы Говерса (последовательность движений, облегчающих вставание с пола), слабость проксимальных мышц, переваливающаяся («утиная») походка, уплотнение ахилловых сухожилий и заметно гипертрофированные мышцы голеней. Уровень креатинкиназы сыворотки крови оказался в 50 раз выше нормы.
Поскольку анамнез и данные медицинского осмотра, включая повышенный уровень креатинкиназы, позволили предположить миопатию, ребенок для дальнейшего обследования направлен в клинику нейрогенетики. Результаты биопсии мышц показали выраженное изменение размера мышечных волокон, некроз волокон, разрастание жировой и соединительной ткани и отсутствие окрашивания на дистрофии. На основании этих результатов ребенку установлен предварительный диагноз мышечной дистрофии Дюшенна, и он тестирован на делеции в гене дистрофина; оказалось, что у него имеется делеция с 45 по 48 экзон.
Прогрессирующая мышечная дистрофия Дюшена – это наследуемое заболевание, которое развивается в раннем возрасте. Характеризуется симметричной атрофией мышц, которая сочетаются с костно-суставными, сердечно-сосудистыми и психическими нарушениями.
Причины
Дистрофия Дюшена возникает по причине поражения гена, отвечающего за выработку дистрофина - белка, необходимого для нормального существования мышечной ткани человека. Это и приводит к развитию заболевания. Если женщина имеет дефектный ген, то она, как правило, не болеет, но передает его своим детям. Мальчики, которые получили этот ген, неизбежно заболевают дистрофией Дюшена. У девочек болезнь развивается очень редко, это может произойти в результате структурных аномалий хромосом.
Симптомы
Основные симптомы заболевания следующие:
- перерождение мышечной ткани;
- некроз некоторых волокон;
- постепенное замещение мышечной ткани жировой и соединительной тканями.
При синдроме Дюшена симптомы проявляются примерно к 18 месяцам жизни. Такие дети отстают в развитии. Первым признаком болезни может стать то, что ребенок к положенному времени не начинает ходить, а все попытки встать на ноги заканчиваются неудачно. Либо ребенок ходит вразвалку и часто падает, в отличие от других детей его возраста. Еще один характерный симптом болезни – попытки ребенка встать из сидячего положения, при этом он пытается поддерживать ноги руками.
Мышцы при прогрессирующей мышечной дистрофии Дюшена всегда атрофируются симметрично. В первую очередь поражаются крупные мышцы нижних конечностей и тазового пояса, далее – мышцы верхней части туловища, а затем – другие основные группы мышц.
Классический признак болезни – это псевдогипертрофия мышц на ногах, то есть икроножных. При этом происходит их ложное увеличение в результате отложения жира и разрастания соединительной ткани.
Псевдогипертрофия может проявляться на дельтовидных, ягодичных мышцах, а также мышцах живота и языка. При надавливании на мышцы больного можно ощутить, что они плотные и абсолютно безболезненные.
При подозрении на мышечную дистрофию проводится диагностика, включающая в себя ряд тестов на определение показателей жизненноважных функций. Доктор назначает проведение следующих исследований:

- Биохимический анализ крови. Здоровая мышечная ткань содержит в себе фермент креатинфосфокиназа. Если у больного присутствует мышечная дистрофия, то уровень этого энзима в крови очень высок.
- Мышечный тест. Посредством проведения электромиографии замеряется скорость проведения в мышцах нервных импульсов. Дети с мышечной дистрофией имеют результаты теста, резко отличающиеся от нормальных.
- Биопсия мышц. В этом случае проводится исследование образца мышечной ткани под микроскопом. При заболевании в ней обнаруживаются отложение жировой ткани и клеточные изменения.
- МРТ . Определяется церебральная атрофия.
- Генетический тест. Раннее обнаружение дефектного гена дает возможность начать лечение как можно раньше и тем самым и отдалить на максимальное время развитие патологий, сохраняя качество жизни больного.
Лечение
Сегодня в медицине нет способов, которые давали бы возможность предотвратить или притормозить прогрессирование мышечной дистрофии. Поэтому основная цель лечения – борьба с осложнениями, поддержание физической активности больного дистрофией и улучшение качества жизни в целом.
Своевременное лечение позволяет замедлить формирование сколиоза, а также позволяет больным двигаться. В некоторых случаях назначается ортопедическое вмешательство. Что касается лекарственной терапии, то часто назначается препарат преднизолон, который способствует замедлению прогрессирования болезни и увеличению мышечной массы.
Профилактика болезни заключается в генетическом консультировании родителей, которое необходимо проводить еще на этапе планирования беременности.
Прогноз
Течение мышечной дистрофии Дюшена - быстро прогрессирующее. Ко второму десятилетию жизни больного человека развиваются значительные двигательные расстройства, которые приводят к невозможности самостоятельного передвижения.
Для последней стадии характерно возникновение проблем с дыхательными мышцами, мышцами глотки и лица. Смерть зачастую наступает от острой сердечной и дыхательной недостаточности, чаще всего это происходит на втором либо третьем десятке жизни.
Ниже можно посмотреть мультфильм с русскоязычными субтитрами, в котором доступно рассказывается о мышечной дистрофии Дюшена: