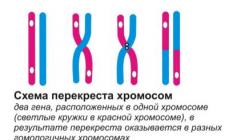
Считается, что при ДГ в хромосомах могут повреждаться гены или локусы по группам, ответственным за половое развитие (J), за нормальное развитие костно-мышечной, сердечно-сосудистой, мочевой и других систем (А), а также за рост (S). В зависимости от сочетания повреждений этих структур возникают разные формы болезни: половой инфантилизм без аномалий развития при высоком росте, карликовость с половым инфантилизмом и аномалиями развития, инфантилизм при малом росте и отсутствии аномалий развития органов и систем и др.
Развитие фолликулов и продукция эстрогенов отсутствуют, что приводит к половому инфантилизму и аменорее. В отдельных случаях допускается возможность сохранения ооцитов и созревания фолликулов до менархе, а затем возникновение преждевременной яичниковой недостаточности и вторичной аменореи. Такое разнообразие форм дисгенезии гонад может быть связано со сроками неблагоприятного воздействия вредных факторов в период дифференцировки или формирования половых желез. Поврежденная ткань гонад в период дифференцировки погибает, замещаясь соединительной тканью, с последующим формированием нейтрального женского фенотипа из-за отсутствия секреции «организатора» мужского типа даже в случаях хромосомной детерминированности мужского типа. Дисгенезию гонад, обусловленную патологией половых хромосом, возникшей на ранних стадиях овогенеза и сперматогенеза, можно считать наследственной.
Половой хроматин при ДГ у большинства больных (80% и более) отсутствует. При этом может иметь место правильный мужской набор хромосом (XY). При положительном половом хроматине отмечается правильный женский набор хромосом (XX). Мозаичный же набор хромосом сопровождается отрицательным или положительным половым хроматином при ДГ.
При ДГ отсутствует должная реакция яичников на гонадотропины, что сопровождается высоким их уровнем, в большей степени ФСГ, а также резкой гипоэстрогенией.
«Чистая» форма дисгенезии гонад характеризуется выраженным половым инфантилизмом у женщин с нормальным или высоким ростом при отсутствии соматических аномалий развития. Впервые описана в 1955 г. Swyer. Больные имеют женский фенотип с диспластическим телосложением по интерсексуальному (увеличение окружности грудной клетки и уменьшении поперечных размеров тела) или евнухоидному (увеличены размеры грудной клетки и ног и уменьшены размеры таза) типам. У них отсутствуют или существенно недоразвиты вторичные половые признаки: скудное оволосение в лобковой и подмышечной областях, недоразвиты молочные железы, наружные половые органы, матка, атрофичная слизистая половых органов. Кариотип у больных чаще всего 46 XX с нормальным или сниженным содержанием полового хроматина. Возможен кариотип 46 XY (синдром Свайера) с резким снижением или отсутствием полового хроматина Уровень гонадотропных гормонов резко увеличен, преимущественно за счет ФСГ (в 8-15 раз) и в меньшей степени за счет ЛГ (в 4-5 раз). Содержание тестостерона и экскреция 17-КС в пределах нормы или с тенденцией к повышению при синдроме Свайера. Патоморфологическая картина характеризуется женским типом половых органов: определяются матка и трубы, на месте яичников - продолговатые белесоватые образования, нередко с единичными примордиальными фолликулами (часто с различными дегенеративными изменениями). При синдроме Свайера в дисгенетичных гонадах нередко образуются гормонально-активные опухоли, что является основанием для удаления гонад при этой патологии (кариотип 45 XY, отрицательный половой хроматин).
Синдром Шерешевского-Тернера (типичная форма ДГ) характеризуется тотальным половым инфантилизмом у женщин с низким ростом и нередко наличием множества соматических аномалий развития. Впервые описан Н.А.Шерешевским в 1926 г. Такие больные рождаются с малой массой тела, отеками рук и ног (синдром Ульриха). В последующем рост у них замедлен (до 50-55% возрастной нормы) и достигает не более 150-155 см. Они характеризуются коренастым телосложением, непропорционально большой грудной клеткой и короткой шеей. Кроме небольшой длины тела, отмечаются другие соматические аномалии: складки на шее, низкая линия оволосения на шее, деформация черепа и локтевых суставов, низко расположенные уши с дефектом хряща ушной раковины, плоскогрудие, дефекты сердечно-сосудистой системы (атрезия клапанов, дефект межпредсердной перегородки, коарктация аорты, стеноз легочной артерии и др.), мочевыводящих путей (подковообразные почки, раздвоение мочеточников), остеопороз и нарушения формы костей, особенно трубчатых. В пубертатном возрасте у больных не появляются вторичные половые признаки: отсутствует оволосение в подмышечных и лобковой областях, неразвиты молочные железы, большие и малые половые губы, матка и влагалище, атрофичная слизистая половых органов. Характерным для синдрома Шерешевского-Тернера является мозаицизм хромосом: X0/XY, Х0/ХХХ, Х0/ХХ и др. При синдроме Шерешевского-Тернера отмечаются существенные гормональные нарушения: высокие уровни ФСГ (в 10-20 раз выше нормы), ЛГ (в 3-4 раза выше нормы), различные аномальные уровни выделения 17-КС (пики повышение - снижение). Матка и трубы в виде рудиментарных структур, рядом с которыми определяются белесоватые соединительнотканные тяжи (рудиментарные гонады). Описываются различные варианты патологического строения гонад: обычная волокнистая ткань; соединительнотканная строма с интерстициальными клетками, дисгенетические гонады с участками корковой зоны и первичными дегенеративными фолликулами, а также с семенными канальцами; рудиментарные мужские гонады. «Смешанная» форма дисгенезии гонад - одна из форм интерсексуализма (гермофродитизма), называется также асимметричной. Выделена в нозологическую форму ДГ Sohval в 1905 г. Характеризуется неопределенным фенотипом, с преобладанием в одних случаях женского, в других мужского. При женском фенотипе имеют место интерсексуальное строение половых органов и гипертрофия клитора При мужском фенотипе в брюшной полости определяется матка с трубами. Гонады у таких больных имеют смешанное строение: с одной стороны, рудиментарная в виде соединительнотканного тяжа гонада, с другой - недоразвитое яичко с клетками типа Сертоли или Лейдига, а также недифференцированные половые железы и матка (гоноциты). Дисгенетическое яичко может располагаться в области яичника с одной стороны, а также в паховом канале или рудиментарной мошонке. Это и послужило поводом к названию «смешанной» формы дисгенезии гонад (или «гонадального дисгенеза»). Больные со «смешанной» формой ДГ имеют высокий рост (хотя встречаются и низкорослые). Соматические аномалии чаще отсутствуют, реже отмечаются такие же, как и при синдроме Шерешевского-Тернера. У больных при ДГ молочные железы неразвиты, оволосение на лобке выражено, отмечается гипертрихоз и низкий тембр голоса. Половой хроматин в большинстве случаев не выявляется, кариотип чаще с нормальным мужским набором (XY) или в виде мозаицизма по типу X0/XY, отмечаются и другие варианты. Считается, что всегда имеет место патология половых хромосом, чем и объясняется нарушение дифференцировки половых желез с функциональной недостаточностью уже в эмбриогенезе. У больных отмечается высокий уровень гонадотропинов с содержанием женских и мужских половых стероидных гормонов на уровне нижней границы мужской нормы. Нередко в дегенеративном яичке развиваются новообразования типа гоноцитомы или гонадобластомы, приводящие к выраженной вирилизации.
В диагностике дисгенезии гонад важное значение имеют общеклинические данные, результаты осмотра и дополнительных методов исследования: УЗИ, определение полового хроматина и кариотипа, лапароскопия и лапаротомия с биопсией гонад, гормональные исследования с проведением функциональных гормональных проб.
Лечение дисгенезии гонад должно проводиться совместно с эндокринологом, генетиком, хирургом, урологом и психологом. Особенно важен такой комплексный подход при выборе тактики и методов лечения при выраженной интерсексуальности половых органов. Основной целью лечения является предотвращение психосексуальных конфликтов. А для этого должны быть по возможности правильно сформулированы или скорригированы нарушенные половые органы и созданы условия (возможности) к половой жизни. Именно это должно быть положено в основу выбора тактики и пола при проведении лечения (а не хромосомный пол). Это подтверждается и современной проявляющейся тенденцией к транссексуализму.
Основные направления при лечении дисгенезии гонад: хирургическая коррекция соматических аномалий развития; мероприятия по коррекции низкого роста; формирование вторичных половых признаков; психотерапевтические воздействия; заместительная гормональная терапия. При проведении лечения должна тщательно соблюдаться деонтология в аспекте правильных взаимоотношений с больными, их родителями и всеми окружающими. Многие вопросы должны обсуждаться и согласовываться только с больными, особенно при достижении ими половозрелого возраста. При синдроме Шерешевского-Тернера, когда диагноз устанавливается сразу после рождения, составляется план и намечаются сроки хирургических корригирующих вмешательств по устранению аномалий соматического развития (пороков костной, сердечно-сосудистой и других систем). Этот этап лечения выполняется детскими хирургами. Следующей важнейшей задачей является стимулирование роста при его задержке, что следует проводить как можно раньше. С этой целью рекомендуются гормоны щитовидной железы, тиреоидные препараты (тиреоидин по 50-100 мг, тиреокомб по 50-100 мг, трийодтиронин по 20-25 мкг, лиотиронин по 20 мкг и др.) и анаболические гормоны (метиландростенолон по 0,1 мг/кг в сутки, нероболил по 1 мл внутримышечно 1 раз в месяц, ретаболил по 1 мл 1 раз в 3 месяца). Они применяются курсами по 4-5 месяцев с 2-3-месячными перерывами. Для стимуляции роста проводится также инсулинотерапия (2-10 ЕД в сутки) курсами по 2 месяца 2 раза в течение года. С этой же целью могут использоваться также факторы роста (соматомедины и факторы роста с инсулиноподобной активностью). Такое лечение проводится до 13-15-летнего возраста пациентов. Этот этап осуществляется под контролем эндокринологов, в процессе которого особенно важной является оценка процессов окостенения.
Своевременное решение вопроса о сроках гонадэктомии является весьма актуальным по следующим соображениям: в связи с потенциальной бластоматозной активностью дисгенетических гонад такие больные составляют группу повышенного риска в онкологическом аспекте; гонадэктомия, по-видимому, должна предшествовать циклической гормональной терапии половыми стероидными гормонами. Гонадэктомия широко используется при синдроме Шерешевского-Тернера и при «смешанной» форме дисгенезии гонад, особенно с кариотипами 45 Х0, при наличии в кариотипе Y-хромосомы или ее фрагментов. Решение этого вопроса осуществляется совместно с урологами и генетиками.
Очередным этапом являются индукция и развитие первичных и вторичных половых признаков. Вначале (с 13-15 лет) назначаются эстрогенные препараты курсами по 20-21 дню с 6-7-дневными перерывами, продолжительностью от 6 месяцев до 2 лет. Преждевременное применение эстрогенов приводит к окостенению эпифизарных костей и задержке роста. Поэтому отдельные авторы рекомендуют начинать их использование с 16-17 лет, что, видимо, не совсем оправданно в плане развития первичных и вторичных половых признаков. Однако половые стероидные гормоны могут назначаться и с 11-12 лет больным с наклонностью к гигантскому росту для его остановки (при чистой и смешанной формах ДГ). Под контролем ТФД, УЗИ и других методов обследования оценивается эффективность эстрогенных соединений (рост матки, молочных желез, состояние слизистых, появление менструальноподобных кровотечений и др.) для перехода на циклическую гормональную терапию. Эстрогенные препараты назначаются парентерально, перорально (фолликулин, этинил-эстрадиол, премарин, синопаузе, димэстрол и др.). Своевременный переход на циклическую гормональную терапию с применением гестагенов для имитации II фазы цикла является необходимым и для профилактики гиперпластических процессов в гормонально зависимых органах (матке и молочных железах). Одновременно рекомендуются комплекс витаминов по фазам цикла (С, В, Е, фолиевая кислота и др.), а также физиотерапевтические процедуры с целью улучшения кровоснабжения органов малого таза, электроанальгезия, иглорефлексотерапия, седативные средства.
При развитии вторичных половых признаков (спонтанных или индуцированных) показана только заместительная циклическая гормональная терапия с имитацией фаз цикла (микрофоллин, прегнин и др.); проводится независимо от формы дисгенезии гонад. Она способствует устранению психопатологических состояний, улучшению общего состояния, нормализации работоспособности и ликвидации других психоэмоциональных, вегетососудистых и обменно-эндокринных нарушений. Терапия также предусматривает профилактику остеопороза и сердечно-сосудистых заболеваний, характерных для гипоэстрогенемии. В процессе гормональной терапии должен проводиться динамический контроль (ТФД, УЗИ, цитология и др.). Осложнения при заместительной гормональной терапии (мастопатия, миома матки, гиперплазия эндометрия) устраняются увеличением гестагенного компонента. Заместительная гормональная терапия проводится на протяжении длительного времени, возможно, в течение всего детородного периода.
Прогноз при дисгенезии гонад в аспекте восстановления специфических женских функций неоднозначен. Таких больных следует относить в группу абсолютного риска по бесплодию. Если в отдельных случаях при чистой форме ДГ и возможно достижение даже овуляторных процессов, то терапия по поводу бесплодия вряд ли должна проводиться, поскольку вероятность рождения здоровых детей у таких больных весьма низкая. В случае возникновения у них беременности должно проводиться тщательное медико-генетическое обследование.
Для развития яичников необходимы две нормальные X-хромосомы. При точечных мутациях генов на X-хромосомах нарушаются самые ранние этапы дифференцировки половых желез. В отсутствие и при крупных аберрациях одной из X-хромосом нарушаются как ранние, так и поздние этапы дифференцировки: ооциты I порядка не вступают в 1-е деление мейоза, не формируются примордиальные фолликулы. Таким образом, строение половых желез при X-хромосомных аберрациях и мутациях генов на X-хромосомах может быть разным. Например, при кариотипе 45,X гонады представлены соединительнотканными тяжами, не содержащими ооцитов I порядка и фолликулов. При других кариотипах яичники недоразвиты, с разным количеством примордиальных фолликулов.
1. Синдром Тернера
а. Этиология . Синдром Тернера в 60% случаев обусловлен моносомией X-хромосомы (кариотип 45,X), в 20% случаев — мозаицизмом и в 20% случаев — аберрацией одной из X-хромосом. Кариотип 45,X вызван нерасхождением половых хромосом в 1-м делении мейоза. Яичники плода с кариотипом 45,X имеют нормальное гистологическое строение до 3-го месяца внутриутробного развития, после чего все или почти все ооциты I порядка и фолликулы дегенерируют. Частота синдрома Тернера с кариотипом 45,X у новорожденных девочек составляет 1:2500. Мозаицизм обусловлен нарушением дробления зиготы. У больных с мозаицизмом встречаются клоны клеток, содержащих две X-хромосомы (45,X/46,XX), X- и Y-хромосомы (45,X/46,XY), либо клоны с полисомией X-хромосомы (например, 45,X/47,XXX). Варианты аберраций X-хромосомы: делеция короткого или длинного плеча (46,X или 46,X соответственно); изохромосома по длинному или по короткому плечу; кольцевая X-хромосома .
б. Клиническая картина. Важнейшие признаки: первичная аменорея и низкорослость. Другие признаки — эпикант, низко расположенные уши, готическое небо, микрогнатия, короткая шея, крыловидные складки на шее, птоз, плоская грудная клетка, О-образное искривление рук (деформация локтевых суставов), лимфатические отеки тыльной поверхности кистей и стоп, укорочение IV и V пястных или предплюсневых костей, гипоплазия ногтей, остеопороз, подковообразная почка, коарктация аорты и цветовая слепота (неспособность различать красный и зеленый цвета). Встречаются варианты синдрома Тернера без множественных аномалий развития.
2. Чистая дисгенезия гонад. У всех больных женский фенотип и нормальный кариотип 46,XX или 46,XY; множественных аномалий развития нет (отсюда слово «чистая» в названии синдрома). Гонады тяжевидные, не содержат ооцитов и фолликулов; матка и маточные трубы недоразвиты. Рост больных нормальный или даже превышает норму (> 170 см). Этот синдром обусловлен точечными мутациями генов на X-хромосоме (при кариотипе 46,XX) или мутацией гена SRY на Y-хромосоме (при кариотипе 46,XY). Распространенность среди женщин — 1:25 000. Чтобы отличить чистую дисгенезию гонад от синдрома Тернера с минимальными соматическими проявлениями, необходимо цитогенетическое исследование. У больных с кариотипом 46,XY может возникнуть дисгерминома или гонадобластома. Симптомы этих опухолей: вирилизация и объемное образование в малом тазу.
3. Смешанная дисгенезия гонад. У больных со смешанной дисгенезией гонад на одной стороне имеется яичко, а на другой — тяжевидная гонада. Кариотип, как правило, — 45,X/46,XY. Направление полового развития зависит от количества клеток с кариотипом 46,XY. Если во внутриутробном периоде яичко функционирует, то формируются наружные половые органы промежуточного типа. Среди причин появления наружных половых органов промежуточного типа смешанная дисгенезия гонад занимает второе место (на первом месте — врожденная гиперплазия коры надпочечников). Почти всегда имеются влагалище, матка и по крайней мере одна маточная труба. Для большинства новорожденных выбирают женский пол воспитания.
4. Обследование
а. Анамнез. Дисгенезию гонад следует заподозрить у любой женщины, обратившейся с жалобами на нарушения менструального цикла, если ее рост менее 150 см. В большинстве случаев при дисгенезии гонад наблюдается первичная аменорея; при легких формах болезни аменорея может быть вторичной.
б. Лабораторная диагностика. Уровни ФСГ и ЛГ в сыворотке повышены.
в. Цитогенетическое исследование необходимо для уточнения диагноза.
г. Если Y-хромосома не обнаружена, определяют антиген H-Y.
д. Аномалии почек и мочевых путей (подковообразная почка, тазовая дистопия почки или удвоение мочеточника) выявляют при экскреторной урографии.
е. Дисгенезия гонад может сочетаться с коарктацией аорты и другими пороками развития сердца и сосудов.
ж. Регулярно исследуют функцию щитовидной железы, поскольку больные с дисгенезией гонад предрасположены к хроническому лимфоцитарному тиреоидиту.
з. До и во время лечения определяют костный возраст по рентгенограммам левой кисти и запястья.
5. Лечение
а. Проводят заместительную терапию эстрогенами. Назначают этинилэстрадиол, 0,02—0,05 мг/сут, или конъюгированные эстрогены, 0,3 мг/сут через день. Считается, что для ускорения роста нужны гораздо меньшие дозы эстрогенов, чем для индукции развития вторичных половых признаков. Поэтому дозы эстрогенов повышают постепенно и подбирают индивидуально таким образом, чтобы имитировать нормальное половое развитие. Назначение минимальных доз эстрогенов в начале лечения позволяет каждой больной достичь максимально возможного роста. Постепенное увеличение доз стимулирует развитие вторичных половых признаков. Впоследствии переходят на циклический режим лечения. Цикл продолжается 28 дней: первые 11 дней принимают только эстрогены; в последующие 10 дней дополнительно назначают медроксипрогестерона ацетат, 5—10 мг/сут; делают перерыв на 7 дней.
Одна из 2000-2500 новорожденных девочек не имеет второй половой хромосомы (как X, так и Y), но имеет структурные реаранжировки Х-хромосомы и/или мозаицизм. Пациенты с кариотипом 45, X составляют около 50% от всех пациентов с нарушениями Х-хромосомы. Было выявлено, что 99% плодов с 45, X не выживают более 28 недель гестации, а 15% от всех выкидышей, произошедших в первом триместре, имеют кариотип 45, X. В 70-80% случаев присутствующая Х-хромосома имеет материнское происхождение.
Отличительными чертами 45, Х-дисгенезии гонад являются разнообразные соматические нарушения, половой инфантилизм в постпубертатном периоде из-за дисгенезии гонад и низкорослость. Пациентки с кариотипом 45, X отличаются и в детском возрасте, обычно за счет лимфатического отека конечностей и характерной кожной складки на шее. В старшем возрасте пациентки обычно узнаваемы по лицу с микрогнатией, эпикантом, выступающими и низко посаженными ушами, рыбоподобным ртом и птозом различной степени выраженности. Грудная клетка воронкообразная, шея короткая, широкая и с крыловидными складками (40% пациенток). К дополнительным нарушениям, ассоциированным с синдромом Тернера, относятся почечная патология (30%), пигментные невусы, вальгусная деформация локтей, врожденный вывих бедра, тенденция к образованию келоидных рубцов, отечность тыла кистей и стоп, укорочение четвертых мета-карпальных и метатарзальных костей, деформация Маделунга (хронический подвывих кисти), сколиоз, рецидивирующий отит, который может привести к потере слуха. Нейросенсорная тугоухость развивается с возрастом и обнаруживается у 61% женщин с синдромом Тернера старше 35 лет. Всем пациенткам следует проводить регулярные скри-нинговые аудио-обследования.
У пациенток с синдромом Тернера отмечается увеличение частоты развития пороков сердечно-сосудистой системы. В различных исследованиях частота встречаемости двустворчатого аортального клапана, выявленного при эхокардиографии, составляет от 9 до 34%. Наличие двустворчатого аортального клапана приводит к росту риска развития подострого бактериального эндокардита и с возрастом приводит к стенозу и/или недостаточности аортального клапана. При скрининговых исследованиях у 10% пациентов выявляется коарктация аорты, которая часто сочетается с крыловидными складками на шее. Дилатация аорты является важным фактором риска развития аневризмы и смерти из-за разрыва аорты. Таким образом, всем пациенткам с синдромом Тернера после установления диагноза следует проводить детальное кардиологическое обследование, а в дальнейшем - периодические эхокардиографические или МРТ-исследования для исключения изменений и/или динамического наблюдения за диаметром аорты и состоянием сердечно-сосудистой системы.
Для всех пациенток обязательными исследованиями являются рутинная внутривенная урография или почечная сонография для исключения хирургически корректируемых пороков развития почек. Наиболее частыми вариантами аномалии почек являются «подковообразные» почки, удвоение почечных лоханок и мочеточников, вторичный гидронефроз из-за лоханочно-мочеточниковой обструкции. Были описаны отсутствие почки или полная почечная эктопия. Внутренние протоки, как и наружные половые органы, у таких пациентов обычно развиты по женскому типу, за исключением редких случаев пациентов с кариотипом 45, X, в котором обнаруживаются Y-аутосомные или Y-X-хромосомные транслокации.
Низкорослость является постоянным признаком синдрома дисгенезии гонад. Средний рост пациентов с кариотипом 45, X составляет 143 см, колеблется в пределах 133-153 см. Обнаруживаемая у пациентов с синдромом дисгенезии гонад низкорослость не обусловлена дефицитом гормона роста или ИРФ-1, половых стероидов или тиреоидных гормонов. Она связана в какой-то степени с гаплоидной недостаточностью генов PHOG/SHOX в псевдо-аутосомных участках Х- и Y-хромосом. Назначение высоких доз рекомбинантного человеческого ГР оказывает выраженный эффект и стимулирует рост.
Дисгенезия гонад является еще одним признаком пациенток с кариотипом 45, X. Обычно гонады представляют собой тяжи и содержат только фиброзную строму, расположенную по спирали. Продольные исследования как базального содержания, так и стимулированной гонадотропин-рилизинг гормоном секреции гонадотропинов у пациентов с дисгенезией гонад показали ослабление отрицательной обратной связи системы гипоталамус-гипофиз у детей и подростков с данным заболеванием. Таким образом, определяемые в плазме крови и в моче концентрации гонадотропинов, особенно ФСГ, повышены в раннем детстве и после 9-10-летнего возраста.
Из-за снижения функции яичников пубертат не наступает самостоятельно, поэтому половой инфантилизм является отличительным признаком этого синдрома. Редко у пациенток с кариотипом 45, X наступает самопроизвольное половое созревание, менархе и беременность.
Другие разнообразные нарушения, ассоциированные с этим синдромом, включают ожирение, неостеопорозные переломы в детском возрасте и остеопорозные переломы во взрослом возрасте, сахарный диабет 2-го типа1, тиреоидит Хашимото, ревматоидный артрит, воспалительные заболевания кишечника, интестинальные телеангиэктазии и кровотечения, гипертензию, колит, ИБС, инсульт и нервную анорексию. Более чем у 50% взрослых пациентов отмечаются повышение активности ферментов печени: подъем трансаминаз, щелочной фос-фатазы и ГГТ. Большинство пациентов с синдромом Тернера имеет нормальное умственное развитие, примерно у 10% отмечается значимое отставание в развитии. Тем не менее у 70% пациенток наблюдаются трудности в обучении, такие как пространственные нарушения, сложности в решении невербальных проблем, невнимательность. Необходимы раннее выявление и коррекция, равно как и психологическая помощь.
Пройти кариотипирование следует фенотипическим женщинам со следующими признаками:
- низкий рост (> 2,5 стандартных отклонений от средних значений) с или без соматической патологии, связанной с синдромом дисгенезии гонад;
- задержка наступления пубертата при увеличении концентрации ФСГ в крови.
Лечение должно быть направлено на максимальное увеличение роста и индукцию развития вторичных половых признаков, а также наступления менархе в соответствии с возрастными нормами. По результатам клинических исследований, было установлено, что в результате лечения рекомбинантным ГР (0,375 мг/кг/нед. в расчете на 7 ежедневных доз) с или без оксандролона (0,0625 мг/кг/сут в течение месяца) отмечается увеличение роста в среднем на 8-10 см через 3-7 недель терапии. Раннее начало лечения ГР (как только диагностировано отставание в росте) дает возможность более длительно проводить лечение и получить большую прибавку роста, что позволяет начинать заместительную терапию эстрогенами в возрасте, соответствующем пубертату. До начала терапии ГР, учитывая ее стоимость, следует провести детальный анализ преимуществ и побочных эффектов лечения, и обсудить их с родителями и ребенком. Carel с соавт. не обнаружили влияния величины роста на различные параметры качества жизни у пациенток с синдромом Тернера, получавших терапию ГР. Синергистического влияния на рост при использовании комбинированной терапии эстрогенами и ГР не отмечено. Тем не менее в последних исследованиях пациентки, получавшие для индуцирования наступления пубертата эстрогены накожно, имели рост выше на 2,1 см по сравнению с теми, кто получал эстрогены перорально. Эти исследования подтвердили, что, как и у взрослых женщин, пероральный прием препаратов эстрогенов снижает содержание ИРФ-1 и угнетает его метаболические эффекты. У пациенток, получавших ГР и достигших нормальных величин роста, и у пациенток с доказанной дисгенезией гонад, отказавшихся от терапии ГР, заместительная терапия эстрогенами обычно начинается после 12-13-летнего возраста.
Для стимуляции развития вторичных половых признаков у девочек с гипогонадизмом все чаще и чаще применяется накожное введение эстрогенов с помощью пластыря или геля, при котором нет прохождения «первичного барьера» через печень. Alkarberg-Lindgren и соавт. обследовали 15 пациенток, применявших пластырь с эстрадиолом в ночное время в течение 12 часов для симуляции суточных ритмов секреции эстрадиола в раннем пубертатном периоде. Разрезая пластырь с 25 мкг эстрадиола на кусочки, содержащие различные дозы препарата, и измеряя сывороточный уровень эстрадиола, авторы заключили, что 0,08-0,12 мкг/кг массы тела - приемлемая начальная доза эстрадиола при трансдермальном применении. Они также рекомендовали целевые уровни эстрадиола в утренние часы, на которые следует ориентироваться для достижения удовлетворительного уровня развития по Tanner. Начальная доза применяется в течение 9 месяцев, затем дозу увеличивают. Цель терапии - достижение среднепубертатного содержания эстрадиола в течение 15 месяцев. Прогестины добавляют к терапии эстрадиолом через 2 года после начала лечения эстрадиолом. Данных о влиянии применения эстрогенового пластыря на размер матки и минеральную плотность костей нет.
Конъюгированные эстрогены (0,3 мг или меньше) или этинилэстрадиол (3-5 мкг) могут применяться перорально 21 день каждого календарного месяца. Затем доза эстрогенов постепенно повышается в течение последующих нескольких лет до 1,25 мг конъюгированных эстрогенов или 10 мкг этинилэстрадиола ежедневно в течение 21 дня месяца. Минимальная доза эстрогенов, которая необходима для лечения, должна быть достаточной для инициации и поддержания роста матки, развития вторичных половых признаков, менархе, а также предупреждения развития остеопороза. После первого года терапии эстрогенами добавляется 5 мг медроксипрогестерона ацетата или похожий прогестин, прием осуществляют с 10-го по 21-й день месяца для наступления физиологической менструации и снижения риска развития карциномы эндометрия, ассоциированного с приемом чистых эстрогенов. Недавно был опубликован алгоритм ведения взрослых пациенток с синдромом Тернера. При первичном приеме следует провести: (1) детальное физикальное обследование, включая измерение роста, веса и АД; (2) общий анализ крови, определение уровня креа-тинина, активности ренина, уровня Т4 и ТТГ, активности печеночных ферментов, содержания антител (патология щитовидной железы и толстой кишки, IgA тканевой трансглутаминазы), липидного профиля, HgA1C, уровня глюкозы и инсулина; (3) DEXA или QTC для оценки костной плотности, эхокардиографию или МРТ (если исследование не было сделано в течение 3 предшествующих лет), аудиограмму и УЗИ органов малого таза для оценки размеров матки и придатков; и, с учетом заболеваемости, фертильности, психического здоровья и медицинского страхования, выделить группы, которым необходимо продолжение эстрогенной заместительной гормональной терапии. При последующих ежегодных визитах следует брать цитологический мазок со слизистой шейки матки и оценивать функцию печени, уровни Т4, ТТГ, липидов, креатинина, HgAlC и ФСГ. С интервалом 3-5 лет проводится DEXA/QTC, аудиограмма и эхокардио-грамма/МРТ для оценки диаметра аорты.
Важно, чтобы врач обсудил диагноз и прогноз с родителями и пациенткой. Информацию необходимо преподносить в соответствии с возрастом пациентки. Возможности восстановления фертильности следует обсуждать в соответствующем возрасте в зависимости от интеллектуального и социального статуса пациентки. Пациентка должна быть уверена в своих сексуальных возможностях и владеть информацией о возможности восстановления фертильности с использованием донорской яйцеклетки и оплодотворения in vitro. После цикла терапии успех имплантации донорской яйцеклетки составляет до 40%; однако лишь 50% беременностей завершаются рождением живого ребенка из-за большого количества выкидышей. Основным противопоказанием является повышенный риск расслоения аорты во время беременности. Все женщины с синдромом Тернера, желающие забеременеть, должны пройти детальное кардиологическое обследование до подсадки донорской яйцеклетки, а при наступлении беременности следует постоянно следить за состоянием сердечно-сосудистой системы. У пациенток 45, X с синдромом Тернера самостоятельное наступление беременности происходит чрезвычайно редко; и при наступлении беременности высок риск невынашивания, а также Х-хромосомной патологии у плода, аналогично риску у лиц с Х-хромосомным мозаицизмом или структурными нарушениями. Последние исследования выявили увеличение смертности и сопутствующее снижение продолжительности жизни из-за патологии коронарных артерий, врожденных пороков сердца, метаболических и эндокринных нарушений. Многие сопутствующие заболевания, обусловливающие снижение продолжительности жизни, можно выявить при детальном медицинском обследовании и провести консервативную терапию или хирургическую коррекцию. Обычная практика назначения эстрогенов 3 недели из 4 была подвергнута критики из-за отсутствия эстрогенов в течение ХА года. Было высказано предположение, что такой режим приема является фактором недостаточного развития матки у женщин с синдромом Тернера. Тем не менее в других исследованиях эта точка зрения не подтвердилась, в одном исследовании было выдвинуто предположение, что матка достигает нормальных размеров в основном при 45, Х/46, XX мозаицизме.
Х-хроматин-положительные варианты синдрома дисгенезии гонад
Характерные соматические клинические проявления, равно как и особенности состояния гонад, могут отмечаться у пациентов с аномалиями структуры Х-хромосомы (делеции или добавления) и мозаицизмом половой хромосомы на фоне набора 45, X. Факты свидетельствуют, что гены как длинного, так и короткого плеч Х-хромосомы определяют половую дифференцировку, тогда как гены короткого плеча Х-хромосомы преимущественно предупреждают развитие низкорослости и соматических нарушений, наблюдаемых у пациентов 45, X. Обычно у пациентов с 45, Х/46, XX мозаицизмом имеется нормальный (а не характерный для 45, X) фенотип и даже может отмечаться нормальное половое развитие и фертильность. У пациентов с дупликацией длинного плеча Х-хромосомы и делецией короткого плеча - с так называемой Xq-изохромосомой - отмечается увеличение частоты развития аутоиммунного тиреодита, сахарного диабета 2-го типа и воспалительных заболеваний кишечника по сравнению с пациентами 45, X. У пациентов с кариотипом 46, Хг (X) может быть отставание в умственном развитии и пороки развития, не всегда связанные с синдромом Тернера. Эти нарушения связаны с недостатком инактивации малого кольца Х-хромосомы и, следовательно, функциональной дисомией генов кольцевой и нормальной Х-хромосом.
Х-хроматин-негативные варианты синдрома дисгенезии гонад
У таких пациентов обычно отмечается мозаицизм 45, X и Y-несущих клеток - 45, Х/46, XY; 45, Х/47, XXY; 45, Х/46, XY/47XXY - или возможны структурные аномалии Y-хромосомы. Пациенты варьируют от фенотипических женщин с симптомами синдрома Тернера до пациентов с неопределенными или полностью вирилизованными гениталиями с несколькими стигмами синдрома Тернера. Варианты дифференцировки гонад колеблются от билатеральных тяжей до билатеральных диспластичных яичек, похожих на «нормальные»; также возможно асимметричное развитие (т. е. тяж с одной стороны и диспластичное яичко или, редко, нормальное яичко с другой стороны), иногда называемое смешанной дисгенезией гонад. Развитие наружных гениталий и внутренних протоков коррелирует со степенью дифференцировки яичек и, предположительно, со способностью яичек плода к секреции антимюллерового гормона и тестостерона.
Риск опухолей половых органов существенно увеличивается у пациентов с мозаицизмом 45, Х/46, XY и билатеральными тяжами; соответственно, при этом синдроме показано профилактическое удаление гонадных тяжей или диспластичных неопустив-шихся яичек. При неоплазии гонад, из которых чаще встречаются гонадобластомы, во время или после пубертата у таких пациентов обычно наблюдается развитие грудных желез. Для скрининга новообразований может применяться сонография, КТ или МРТ органов малого таза. Гонадобластомы кальцифицируются, и поэтому визуализируются даже при обычной рентгенографии брюшной полости.
Диагностика мозаицизма 45, Х/46, XY основывается на обнаружении клеток 45, X и 46, XY в крови, коже или гонадах. При некоторых вариантах мозаицизма сигнальная хромосома обнаруживается как цитогенетически не различимая - X или Y. В таких случаях используют флуоресцентную гибридизацию in situ (FISH) или молекулярный анализ с Х- и Y-специфичными зондами для определения происхождения сигнальной хромосомы, так как гонадобластомы встречаются у пациентов с отсутствующей Y-хромосомы - даже при деле-ции гена SRY. Решение относительно пола должно основываться на возрасте, в котором был поставлен диагноз, и возможности нормального функционирования наружных половых органов. Большинство пациентов с мозаицизмом 45, Х/46, XY, диагностированным при амниоцентезе, имеют нормальные мужские гениталии и яички с нормальным гистологическим строением. Гениталии неопределенного строения описаны у пациентов с ошибочно диагностированным мозаицизмом 45, Х/46, XY. Мы наблюдали низкорослого 30-летнего мужчину с установленным 45, Х/46, XY мозаицизмом, имевшего нормальные мужские половые органы и фертильность.
У фенотипических женщин с мозаицизмом 45, Х/46, XY, с предопределенным женским полом, дисгенетичные гонады должны быть удалены из-за риска малигнизации. Применение эстрогеновой терапии начинают с пубертата, как и у пациентов с кариотипом 45, X. У детей с таким кариотипом и предопределенным мужским полом все ткани половых органов, за исключением функционально и гистологически нормальных и находящихся в мошонке, должны быть удалены. При гипоспадии проводится ее коррекция. В пубертатном периоде, в зависимости от сохранности функции гонад, показано начало заместительной терапии андрогенами в дозах, аналогичных прописываемым пациентам с неполной формой XY-дисгенезии гонад. Пациентам с яичками, опушенными в мошонку, показано клиническое обследование и УЗИ. Биопсия гонад показана в постпубертатном периоде для выявления возможной карциномы in situ, предракового состояния. Если выявлено заболевание, показана лучевая терапия в низких дозах или гонадэктомия после извлечения спермы.
В подростковом возрасте проводится биопсия гонад детям с мозаицизмом 45, Х/46, XY, имеющим нормальные гениталии и сохранные яички по данным исследования АМГ, ингибинов А и В, содержания гонадотропинов и МРТ органов малого таза. Если при биопсии и УЗИ не выявляется карцинома in situ, следующую биопсию рекомендуется проводить в 20-летнем возрасте. Вопрос о риске озлокачествления тканей гонад у мужчин с мозаицизмом 45, Х/46, XY, имеющих нормальные мужские половые органы и гистологически и функционально нормальные яички, опустившиеся в мошонку, все еще остается открытым, хотя мы предполагаем, что он не выше, чем у мужчин с кариотипом 46, XY.
Дисгенетические 46, XX и 46, XY НПР (46, XX и 46, XY дисгенезия гонад)
Термин дисгенезии XX и XY применяется у пациентов с кариотипом 46, XX и 46, XY, имеющих билатеральные гонадные тяжи, женский фенотип и не имеющих соматических признаков синдрома Тернера. В возрасте периода пубертата у таких пациенток отмечается половой инфантилизм, постка-страционные концентрации гонадотропинов в крови и в моче, нормальный или высокий рост, евнухоидные пропорции тела.
Дисгенетические 46, XX НПР (46, XX дисгенезия гонад)
Дифференцировка яичников, как и яичек, зависит от ряда аутосомных генов, включая WT1, SF-1, DMRT1, WNT4, FOXL2, и фоллистатина для нормального органогенеза. К тому же, в отличие от яичек, для нормального развития и функционирования яичники нуждаются в наличии стволовых клеток. Таким образом, к ХХ-дисгенезии гонад могут привести мутации в генах аутосом и Х-хромосомы, происходящие, к примеру, при дифференцировке и развитии стволовых клеток, миграции в закладку гонад и мейозе в ооцитах. Распространение CAG повторяющегося участка на непостоянном Х-локусе может вызвать повышенное истощение ооцита. Гаплоидная недостаточность FOXL2 - гена, расположенного на хромосоме 3q23 - является причиной аутосомно доминантного блефарофимоз-птоз-эпикант инверсивного синдрома (БПЭС типа 1) и ХХ-дисгенезии гонад. Был описан женский фенотип с мутациями в FOXL2, но без БПЭС.
Согласно полученным в Финляндии данным, семейные и спорадические случаи ХХ-дисгенезии гонад, встречаются с частотой 1:8300 женщин. Анализ генеалогического древа пациентов с семейными случаями заболевания позволил выявить аутосомно-рецессивный путь наследования. При анализе семейных случаев заболевания в Финляндии был выявлен участок хромосомы 2р, ответственный за развитие ХХ-дисгенезии гонад у женщин; это ген, кодирующий рецептор ФСГ, локализованный на хромосоме 2р. У пациентов с ХХ-дисгенезией гонад была выявлена мутация 7-го экзона этого гена. Мутация обнаруживалась во внеклеточном лиганд-связывающем домене рецептора ФСГ, за счет чего снижалась связывающая способность и, соответственно, передача сигналов рецептором. В период полового созревания это приводит к различным нарушениям функций яичников, вплоть до «стрековых гонад» с гипергонадотропным гипогонадиз-мом у некоторых девушек. В ряде исследований Западной Европы и США у женщин с 46, XX дисгенезией гонад не выявлялись мутации гена рецептора ФСГ, что подтверждает редкую встречаемость данной мутации как причины ХХ-дисгенезии гонад вне Финляндии. Гомозиготные мужчины с такой мутацией фенотипически здоровы, однако сперматогенез может быть как нормальным, так и полностью отсутствовать.
Исследования семейных когорт подтвердили гетерогенность патогенеза данного синдрома. Описаны сиблинги - оба с агенезией гонад, но один с кариотипом 46, XX, а другой с кариотипом 46, XY; это подтверждает участие аутосомных генов в формировании заболевания. Скорее всего, у этих пациентов не было дефекта рецептора ФСГ, если учесть развитие нормального мужского фенотипа XY при мутации такого типа; возможно, мутация была в аутосомно-рецессивном гене, участвующем в детерминации гонад. В одной семье у 4 женщин была выявлена наследственная интерстициальная делеция длинного плеча Х-хромосомы, включавшей участок q21-q27. Этот участок предположительно содержит ген или гены, регулирующие развитие и функционирование яичников. В трех семьях была отмечена ассоциация ХХ-дисгенезии гонад с нейросенсорной тугоухостью. В некоторых пораженных группах сиблингов отмечался широкий спектр клинических проявлений: от нормального развития молочных желез и наступления менархе до вторичной аменореи, что отражало разнообразные степени нарушения овариальной функции.
Диагноз 46, XX дисгенезии гонад должен быть исключен у фенотипических женщин с половым инфантилизмом и нормальными мюллеровскими структурами, без соматических стигм синдрома дисгенезии гонад (синдрома Тернера). При кариотипировании выявляются только 46, XX клетки. Как и при синдроме Тернера, концентрации гонадотропинов повышены, а эстрогенов - снижены, при этом на УЗИ или МРТ выявляются гонадные тяжи или недоразвитые яичники. Лечение заключается в применении заместительной эстрогено-прогестероновой терапии в циклическом режиме, как было описано выше для пациентов с синдромом Тернера.
Спорадические случаи ХХ-дисгенезии гонад, аналогично семейным формам, могут быть представлены гетерогенной с патогенетической точки зрения группой пациентов. ХХ-дисгенезию гонад следует дифференцировать от овариальной недостаточности вследствие: лучевой или химиотерапии при лечении злокачественных опухолей у детей, перенесенных инфекций (таких как эпидемический паротит), циркулирующих антител к рецепторам гонадотропинов, биологически неактивного ФСГ, нечувствительности яичников к гонадотропинам, галактоземии, а также нарушений синтеза стероидов (эстрогенов) - например, недостаточность аро-матазы. В последнем случае при проведении УЗИ или МРТ выявляются поликистозные яичники.
Дисгенетические 46, XY НПР (46, XY дисгенезия гонад)
46, XY дисгенезия гонад возникает как спорадически, так и в рамках семейных случаев. Пациенты с полной формой этого синдрома характеризуются женскими наружными половыми органами, нормальным или высоким ростом, билатеральными стрекгонадами, наличием производных мюллеровых протоков, половым инфантилизмом, евнухоидной внешностью и кариотипом 46, XY. Достаточно часто встречается гипертрофия клитора и, как в семейных, так и в спорадических случаях, возможны варианты проявлений от полной картины синдрома до двойственного развития наружных половых органов. Фенотипически картина полного и неполного синдрома XY-дисгенезии гонад зависит от степени дифференцировки ткани яичек, возможностей функционирования яичек плода, секреции тестостерона и антимюллерового гормона. В раннем детском возрасте и в период полового созревания концентрации гонадотропинов значительно повышены.
При анализе спорадических и семейных случаев 46, XY дисгенезии гонад было обнаружено, что у 15-20% пациентов имеются мутации в HMG участке гена SRY, участвующего в связывании или изгибании ДНК с помощью белка SRY, или мутации внутриядерных сигнальных молекул - калмо-дулина или считывающего домена гена SRY. У большинства пациентов с выявленными мутациями отмечался «полный» синдром дисгенезии гонад. У пациентов с грубой делецией короткого плеча Y-хромосомы, помимо дисгенезии гонад, могут присутствовать стигмы синдрома Тернера. Мутации в HMG участке гена SRY встречаются у здоровых 46, XY отцов «дочерей» с 46, XY дисгенезией гонад. Такие семейные когорты предполагают, что мутации и изменчивость генов могут повредить либо уровень, либо время начала экспрессии SRY, что определяет соответственно либо нормальную, либо нарушенную дифференцировку яичек. У лиц без выявленных нарушений гена SR Y причинами заболевания могут быть мутации вне HMG участка гена SRY, а также в Х-сцепленных или аутосомных генах, ответственных за окончательную дифференцировку и функционирование яичек.
Более чем у 20 пациентов с 46, XY дисгенезией гонад была выявлена дупликация Хр21.2 -> р22.11 участка Х-хромосомы. В этом участке находится ген DAX1. Делеция или мутация DAX1 у мужчин вызывает гипоплазию надпочечников и гипогона-дотропный гипогонадизм. Тот факт, что мужчины с гипоплазией надпочечников и гипогонадотроп-ным гипогонадизмом имеют нормальную половую дифференцировку подтверждает, что DAX1 не является необходимым для половой дифференцировки человека, но тем не менее влияет на дифференцировку яичек.
В литературе описаны женщины с надпочечни-ковой недостаточностью, у которых отсутствуют гонады и мюллеровские структуры на фоне кариотипа 46, XY и гетеро- и гомозиготных мутаций в SF-1. В последнее время был существенно расширен фе-нотипический спектр мутаций SF-1, и в него вошли пациенты с дисгенезией гонад и с кариотипом 46, XY с нормальной функцией надпочечников. В одном из исследований было установлено, что трое из 6 пациентов с кариотипом 46, XY с полной дисгенезией гонад имели гомозиготные мутации в гене DHH. В исследованиях на лабораторных животных было подтверждено, что в тестикулах зародышей мышей Dhh экспрессируется сразу после Sry, регулируя формирование семенных канатиков и дифференцировку клеток Лейдига.
Из-за мутации одной аллели SRY-связанных генов SOX9 на 17-й хромосоме возникает XY-дис-генезия гонад, ассоциированная с кампомелической дисплазией. К тому же XY-дисгенезия гонад может быть связана с 9р- (DMRT1) и 10q- делениями, а также с дупликацией 1р31-35 (WNT4). Причиной XY-дисгенезии гонад в сочетании с аномалиями развития почек (т.е. синдрома Дениса-Драша, Фрейзера и WAGR-синдрома) являются мутации и делеции в гене-репрессоре опухоли Вильмса WT1.
Лечение фенотипических женщин с 46, XY дисгенезией гонад включает профилактическую гонадэкто-мию после установления диагноза и заместительную эстрогенную терапию в пубертатном периоде. При неполной форме XY-дисгенезии гонад в детском возрасте рекомендуется установление мужского пола и лечение заместительной терапией тестостероном для увеличения размеров полового члена. Необходимо рассматривать вопрос о профилактической гонадэктомии, так как фертильность не сохранена (гонадотропины всегда, без исключения, повышены, концентрации АМГ и ингибина В низкие или неопределяемые), к тому же имеется повышенный риск злокачественной трансформации недоразвитых гонад. При сохранении гонад следует проводить их биопсию в пре- и постпубертатном возрасте для выявления начальных злокачественных образований (карцинома in situ). При ее выявлении после извлечения спермы показана гонадэктомия или местная лучевая терапия низкими дозами. У пациентов с мужской идентификацией пола рекомендуется проведение протезирования яичек во время гонадэктомии и поддерживающей терапии андроге-нами в пубертатном периоде. Применяют масляную форму тестостерона энантата (или другого пролонгированного эфира тестостерона), начиная с 50 мг внутримышечно каждые 4 недели, после достижения костного возраста 14,5 лет постепенно увеличивают дозу до полной заместительной - 200 мг внутримышечно каждые 2 недели.
› Отсутствие полового развития (дисгенезия гонад)
Отсутствие полового развития (дисгенезия гонад)
Дисгенезия гонад (ДГ) - врожденный дефект развития половых желез или их полное отсутствие, который чаще обусловлен хромосомными аномалиями (количественная или структурная патология половых хромосом). У больных встречается неполный набор хромосом (45Х вместо 45 ХУ), мозаицизм (ХО/ХХ, XO/XY и др.), дефект короткого плеча Х-хромосомы и т.д. При этом вследствие подавления мейоза яичники не развиваются (на их месте образуются лентовидные полоски белесоватой ткани), а ооциты из них исчезают еще во внутриутробном развитии или сразу после рождения.
Считается, что при ДГ
в хромосомах могут повреждаться гены или локусы по группам, ответственным за половое развитие (J), за нормальное развитие костно-мышечной, сердечно-сосудистой,
мочевой и других систем (А), а также за рост (S). В зависимости от сочетания повреждений этих структур возникают разные формы болезни: половой инфантилизм без аномалий развития при высоком росте, карликовость с половым инфантилизмом и аномалиями развития, инфантилизм при малом росте и отсутствии аномалий развития органов и систем и др.
Развитие фолликулов и продукция эстрогенов отсутствуют, что приводит к половому инфантилизму и аменорее. В отдельных случаях допускается возможность сохранения ооцитов и созревания фолликулов до менархе, а затем возникновение преждевременной яичниковой недостаточности и вторичной аменореи. Такое разнообразие форм дисгенезии гонад может быть связано со сроками неблагоприятного воздействия вредных факторов в период дифференцировки или формирования половых желез. Поврежденная ткань гонад в период дифференцировки погибает, замещаясь соединительной тканью, с последующим формированием нейтрального женского фенотипа из-за отсутствия секреции «организатора» мужского типа даже в случаях хромосомной детерминированности мужского типа. Дисгенезию гонад, обусловленную патологией половых хромосом, возникшей на ранних стадиях овогенеза и сперматогенеза, можно считать наследственной.
Половой хроматин
при ДГ у большинства больных (80% и более) отсутствует. При этом может иметь место правильный мужской набор хромосом (XY). При положительном половом хроматине отмечается правильный женский набор хромосом (XX). Мозаичный же набор хромосом сопровождается отрицательным или положительным половым хроматином при ДГ.
При ДГ отсутствует должная реакция яичников на гонадотропины, что сопровождается высоким их уровнем, в большей степени ФСГ, а также резкой гипоэстрогенией.
«Чистая» форма дисгенезии гонад характеризуется выраженным половым инфантилизмом у женщин с нормальным или высоким ростом при отсутствии соматических аномалий развития. Впервые описана в 195 5 г. Swyer. Больные имеют женский фенотип с диспластическим телосложением по интерсексуальному (увеличение окружности грудной клетки и уменьшении поперечных размеров тела) или евнухоидному (увеличены размеры грудной клетки и ног и уменьшены размеры таза) типам. У них отсутствуют или существенно недоразвиты вторичные половые признаки: скудное оволосение в лобковой и подмышечной областях, недоразвиты молочные железы, наружные половые органы, матка, атрофичная слизистая половых органов.
Кариотип у больных чаще всего 46 XX с нормальным или сниженным содержанием полового хроматина. Возможен кариотип 4б XY (синдром Свайера) с резким снижением или отсутствием полового хроматина. Уровень гонадотропных гормонов резко увеличен, преимущественно за счет ФСГ (в 8- 15 раз) и в меньшей степени за счет ЛГ (в 4-5 раз).
Содержание тестостерона и экскреция 17-КС в пределах нормы или с тенденцией к повышению при синдроме Свайера. Патоморфологическая картина характеризуется женским типом половых органов: определяются матка и трубы, на месте яичников - продолговатые белесоватые образования, нередко с единичными примордиальными фолликулами (часто с различными дегенеративными изменениями). При синдроме Свайера в дисгенетичных гонадах нередко образуются гормональноактивные опухоли, что является основанием для удаления гонад при этой патологии (кариотип 4 5 XY, отрицательный половой хроматин).
Синдром Шерешевского-Тернера (типичная форма ДГ) характеризуется тотальным половым инфантилизмом у женщин с низким ростом и нередко наличием множества соматических аномалий развития. Впервые описан Н АШерешевским в 1926 г. Такие больные рождаются с малой массой тела, отеками рук и ног (синдром Ульриха). В последующем рост у них замедлен (до 50-55% возрастной нормы) и достигает не более 150-155 см.
Они характеризуются коренастым телосложением, непропорционально большой грудной клеткой и короткой шеей. Кроме небольшой длины тела, отмечаются другие соматические аномалии: складки на шее, низкая линия оволосения на шее, деформация черепа и локтевых суставов, низкорасположенные уши с дефектом хряща ушной раковины, плоскогрудие, дефекты сердечно-сосудистой системы (атрезия клапанов, дефект межпредсердной перегородки, коарктация аорты, стеноз легочной артерии и др.), мочевыводящих путей (подковообразные почки, раздвоение мочеточников), остеопороз и нарушения формы костей, особенно трубчатых.
В пубертатном возрасте у больных не появляются вторичные половые признаки: отсутствует оволосение в подмышечных и лобковой областях, неразвиты молочные железы, большие и малые половые губы, матка и влагалище, атрофичная слизистая половых органов. Характерным для синдрома Шерешевского-Тернера является мозаицизм хромосом: X0/XY, ХО/ХХХ, ХО/ХХ и др. При синдроме Шерешевского-Тернера отмечаются существенные гормональные нарушения: высокие уровни ФСГ (в 10-20 раз выше нормы), ЛГ (в 3-4 раза выше нормы), различные аномальные уровни выделения 17-КС (пики повышение - снижение).
Матка и трубы в виде рудиментарных структур, рядом с которыми определяются белесоватые соединительнотканные тяжи (рудиментарные гонады). Описываются различные варианты патологического строения гонад: обычная волокнистая ткань; соединительнотканная строма с интерстициальными клетками, дисгенетические гонады с участками корковой зоны и первичными дегенеративными фолликулами, а также с семенными канальцами; рудиментарные мужские гонады.
«Смешанная» форма дисгенезии гонад - одна из форм интерсексуализма (гермофродитизма), называется также асимметричной. Выделена в нозологическую форму nTSohval в 1905 г.Характеризуется неопределенным фенотипом, с преобладанием в одних случаях женского, в других мужского. При женском фенотипе имеют место интерсексуальное строение половых органов и гипертрофия клитора. При мужском фенотипе в брюшной полости определяется матка с трубами.
Гонады у таких больных имеют смешанное строение: с одной стороны, рудиментарная в виде соединительнотканного тяжа гонада, с другой - недоразвитое яичко с клетками типа Сертоли или Лейдига, а также недифференцированные половые железы и матка (гоноциты). Дисгенетическое яичко может располагаться в области яичника с одной стороны, а также в паховом канале или рудиментарной мошонке. Это и послужило поводом к названию «смешанной» формы дисгенезии гонад (или «гонадального дисгенеза»).
Больные со «смешанной» формой ДГ имеют высокий рост (хотя встречаются и низкорослые). Соматические аномалии чаще отсутствуют, реже отмечаются такие же, как и при синдроме Шерешевского-Тернера. У больных при ДГ молочные железы неразвиты, оволосение на лобке выражено, отмечается гипертрихоз и низкий тембр голоса. Половой хроматин в большинстве случаев не выявляется, кариотип чаще с нормальным мужским набором (XY) или в виде мозаицизма по типу X0/XY, отмечаются и другие варианты.
Считается, что всегда имеет место патология половых хромосом, чем и объясняется нарушение дифференцировки половых желез с функциональной недостаточностью уже в эмбриогенезе. У больных отмечается высокий уровень гонадотропинов с содержанием женских и мужских половых стероидных гормонов на уровне нижней границы мужской нормы. Нередко в дегенеративном яичке развиваются новообразования типа гоноцитомы или гонадобластомы, приводящие к выраженной вирилизации.
В диагностике дисгенезии гонад важное значение имеют общеклинические данные, результаты осмотра и дополнительных методов исследования: УЗИ, определение полового хроматина и кариотипа, лапароскопия и лапаротомия с биопсией гонад, гормональные исследования с проведением функциональных гормональных проб.
Лечение дисгенезии гонад.
Лечение дисгенезии гонад должно проводиться совместно с эндокринологом, генетиком, хирургом, урологом и психологом. Особенно важен такой комплексный подход при выборе тактики и методов лечения при выраженной интерсексуальности половых органов. Основной целью лечения является предотвращение психосексуальных конфликтов. А для этого должны быть по возможности правильно сформулированы или скорригированы нарушенные половые органы и созданы условия (возможности) к половойжизни. Именно это должно быть положено в основу выбора тактики и пола при проведении лечения (а не хромосомный пол). Это подтверждается и современной проявляющейся тенденцией к транссексуализму .
Основные направления при лечении дисгенезии гонад: хирургическая коррекция соматических аномалий развития; мероприятия по коррекции низкого роста; формирование вторичных половых признаков; психотерапевтические воздействия; заместительная гормональная терапия. При проведении лечения должна тщательно соблюдаться деонтология в аспекте правильных взаимоотношений с больными, их родителями и всеми окружающими. Многие вопросы должны обсуждаться и согласовываться только с больными, особенно при достижении ими половозрелого возраста.
При синдроме Шерешевского-Тернера , когда диагноз устанавливается сразу после рождения, составляется план и намечаются сроки хирургических корригирующих вмешательств по устранению аномалий соматического развития (пороков костной, сердечно-сосудистой и других систем). Этот этап лечения выполняется детскими хирургами. Следующей важнейшей задачей является стимулирование роста при его задержке, что следует проводить как можно раньше. С этой целью рекомендуются гормоны щитовидной железы, тиреоидные препараты (тиреоидин по 50- 100 мг, тиреокомб"по 50- 100 мг, трийодтиронин по 20-25 мкг, лиотиронин по 20 мкг и др.) и анаболические гормоны (метилан-дростенатюн по 0,1 мг/кг в сутки, неробалил по 1 мл внутримышечно 1 раз в месяц,ретаболил по 1 мл 1 раз в 3 месяца). Они применяются курсами по 4-5 месяцев с 2-3-месячными перерывами.
Для стимуляции роста проводится также инсулинотерапия (2-10 ЕД в
сутки) курсами по 2 месяца 2 раза в течение года. С этой же целью могут использоваться также факторы роста (соматомедины и факторы роста с инсулиноподобной активностью). Такое лечение проводится до 13-15-летнего возраста пациентов. Этот этап осуществляется под контролем эндокринологов, в процессе которого особенно важной является оценка процессов окостенения.
Своевременное решение вопроса о сроках гонаджтомии является весьма актуальным по следующим соображениям: в связи с потенциальной бластоматозной активностью дисгенетических гонад такие больные составляют группу повышенного риска в онкологическом аспекте; гонадэктомия, по-видимому, должна предшествовать циклической гормональной терапии половыми стероидными гормонами. Гонадэктомия широко используется при синдроме Шерешевского- Тернера и при «смешанной» форме дисгенезии гонад, особенно с кариотипами 45 Х0, при наличии в кариотипе Y-хромосомы или ее фрагментов. Решение этого вопроса осуществляется совместно с урологами и генетиками.
Очередным этапом являются индукция и развитие первичных и вторичных половых признаков. Вначале (с 13- 15 лет) назначаются эстрогенные препараты курсами по 20-21 дню с б-7-дневными перерывами, продолжительностью от б месяцев до 2 лет. Преждевременное применение эстрогенов приводит к окостенению эпифизарных костей и задержке роста. Поэтому отдельные авторы рекомендуют начинать их использование с 1 б- 17 лет, что, видимо, не совсем оправданно в плане развития первичных и вторичных половых признаков.
Однако половые стероидные гормоны могут назначаться и с 11 - 12 лет больным с наклонностью к гигантскому росту для его остановки (при чистой и смешанной формах ДГ). Под контролем ТФД, УЗИ и других методов обследования оценивается эффективность эстрогенных соединений (рост матки, молочных желез, состояние слизистых, появление менструальноподобных кровотечений и др.) для перехода на циклическую гормональную терапию. Эстрогенные препараты назначаются парентерально, перорально (фолликулин, этинил-эстрадиол, премарин, синопаузе, димэстрол и др.).
Своевременный переход на циклическую гормональную терапию с применением гестагенов для имитации II фазы цикла является необходимым и для профилактики гиперпластических процессов в гормонально зависимых органах (матке и молочных железах). Одновременно рекомендуются комплекс витаминов по фазам цикла (С, В, Е, фолиевая кислота и др.), а также физиотерапевтические процедуры с целью улучшения кровоснабжения органов малого таза, электроанальгезия, иглорефлексотерапия, седативные средства.
При развитии вторичных половых признаков (спонтанных или индуцированных) показана только заместительная циклическая гормональная терапия с имитацией фаз цикла (микрофоллин, прегнин и др.); проводится независимо от формы дисгенезии гонад. Она способствуетустранению психопатологических состояний, улучшению общего состояния, нормализации работоспособности и ликвидации других психоэмоциональных, вегетососудистых и обменно-эндокринных нарушений. Терапия также предусматривает профилактику остеопороза и сердечно-сосудистых заболеваний, характерных для гипоэстрогенемии.
В процессе гормональной терапии должен проводиться динамический контроль (ТФД, УЗИ, цитология и др.). Осложнения при заместительной гормональной терапии (мастопатия, миома матки, гиперплазия эндометрия) устраняются увеличением гестагенного компонента. Заместительная гормональная терапия проводится на протяжении длительного времени, возможно, в течение всего детородного периода.
Прогноз при дисгенезии гонад в аспекте восстановления специфических женских функций неоднозначен. Таких больных следует относить в группу абсолютного риска по бесплодию. Если в отдельных случаях при чистой форме ДГ и возможно достижение даже овуляторных процессов, то терапия по поводу бесплодия вряд ли должна проводиться, поскольку вероятность рождения здоровых детей у таких больных весьма низкая. В случае возникновения у них беременности должно проводиться тщательное медикогенетическое обследование.
Частота дисгенезии гонад 1:2500 новорожденных девочек. Диагноз ставят либо сразу после рождения по сопутствующим врожденным порокам, либо, что чаще, в пубертатном возрасте, когда врожденным аномалиям сопутствует аменорея. Дисгенезия гонад наиболее частая причина (30–43%) среди всех форм первичной аменореи на фоне отсутствия вторичных половых признаков.

Этиология Причиной возникновения дисгенезии гонад являются хромосомные и генетические аномалии. Происходит мутация генов, принимающих участие в дифференцировке организма по мужскому типу. В результате нарушения гонадогенеза в эмбриональном периоде половые железы закладываются как соединительнотканные тяжи или недифференцированные гонады с наличием элементов мужских половых желёз (клетки Сертоли, клетки Лейдига, тубулярные структуры). В отсутствие влияния антимюллерова гормона (MIS субстанции) и андрогенов развитие внутренних и наружных половых органов происходит по женскому типу.

Патогенез Для развития яичников необходимо наличие двух половых Х хромосом, т.е. женский кариотип 46,ХХ. В ходе мейотического деления половых клеток возможно возникновение аномального набора половых хромосом. При слиянии таких половых клеток в оплодотворённую яйцеклетку попадает патологический набор хромосом. Хромосомные дефекты могут быть количественными: отсутствие одной хромосомы (моносомия 45,Х), удвоение или утроение числа хромосом (47,ХХХ или 47,ХХY полисомия). Возможно образование мозаичных наборов хромосом, когда клоны клеток имеют различный набор хромосом. В результате неправильного морфофункционального развития яичники не могут продуцировать половые стероиды. Дефицит эстрогенов по принципу обратной связи приводит к повышению синтеза гонадотропинов, поэтому эта аменорея гипергонадотропная. Кроме того, в Х-хромосоме находятся гены, детерминирующие не только половое, но и соматическое развитие.

КЛАССИФИКАЦИЯ Выделяют: Типичную форму (синдром Шерешевсого– Тернера) кариотип 45,Х; Стёртую форму кариотип имеет мозаичный характер, 45,Х/46,ХХ; Чистую форму (синдром Свайера) кариотип 46,ХX или 46 XY. Смешанную форму мозаичный кариотип с обязательным присутствием Yхромосомы или её участка (наиболее часто встречается кариотип 45,Х/46,ХY);

Синдром Шерешевского- Тёрнера (типичная форма дисгенезия гонад) характеризуется широким диапазоном хромосомных аномалий. Больные имеют коренастое телосложение и неправильную осанку, непропорционально большую щитообразную грудную клетку с широко расставленными сосками неразвитых молочных желёз, вальгусную девиацию локтевых и коленных суставов, аплазию фаланг, множественные родимые пятна или витилиго, гипоплазию IV и V фаланг и ногтей. Нередко встречают короткую «шею сфинкса» с крыловидными складками кожи (листовидная шея), идущими от ушей до плечевого отростка, и низкую линию роста волос на шее. Для больных характерны такие изменения костей лицевого черепа, как рыбий рот, птичий профиль за счёт микро и ретрогнатии, деформация зубов. Черты лица изменены за счёт косоглазия, эпикантуса, птоза и деформации ушных раковин. Возможны нарушение слуха, врождённые пороки сердца, аорты и мочевыделительных органов, встречают гипотиреоз, аутоиммунный тиреоидит и сахарный диабет. Определяют недоразвитие вторичных половых признаков, генитальный инфантилизм.


Стертая форма При стёртых формах наиболее часто выявляется мозаичный характер кариотипа 45 ХО/46ХХ. При преобладании клона 45ХО больные ближе по внешнему виду к клинической картине синдрома Шерешевского– Тернера. Превалирование нормального клеточного клона 46ХХ сглаживает соматические признаки типичной формы ДГ. У больных реже наблюдается низкий рост, может быть недостаточное, но спонтанное развитие вторичных половых органов при наличии первичной аменореи. Своевременное наступление менструаций бывает у 20% больных, а у 10% отмечают относительно регулярные менструации в течение 10 лет после менархе, которые потом переходят в олигоменорею и вторичную аменорею.При осмотре наружные половые органы гипопластичны.

Клиническая картина Чистая форма. У пациенток с чистой формой дисгенезии гонад, или синдромом Свайера, при резко выраженном половом инфантилизме отсутствуют соматические аномалии развития. Рост обычный или ниже среднего, молочные железы не развиты, половое оволосение скудное или отсутствует. Кариотип у больных чаще всего 46, ХХ, 46, XY При обследовании наружные половые органы, влагалище и матка недоразвиты, яичники рудиментарные, типична первичная аменорея.

Диагностика Анамнез Выясняют наличие стигм наследственных и врождённых синдромов и особенностей полового созревания обоих родителей и ближайших родственников (I и II степени родства). Матери девочек с дисгенезией гонад нередко указывают на воздействие во время беременности физических и химических вредностей, высокую или частую лучевую нагрузку (рентгеновское, сверхвысокочастотное, лазерное и ультразвуковое излучение), обменные и гормональные нарушения, интоксикации на фоне приёма эмбриотоксичных препаратов и наркотических веществ, острые инфекционные заболевания, особенно вирусной природы. До пубертатного возраста развитие ребёнка с XY дисгенезией гонад не отличается от сверстников. В пубертатном возрасте, несмотря на своевременное половое оволосение, развитие молочных желёз отсутствует, менархе не возникает.

Лабораторные исследования. Наиболее информативно определение гормонов в сыворотке крови, для которых характерно резкое повышение уровня гонадотропинов (ЛГ, ФСГ), сниженные концентрации эстрадиола. Генетическое обследование включает определение полового хроматина в буккальных мазках и кариотипа, при котором выявляют отсутствие полового хроматина и типичный для той или иной формы кариотип. Гормональная проба с гестагенами отрицательная, что доказывает выраженный дефицит эстрогенов; проба с эстрогенами и гестагенами положительная, что исключает маточную форму аменореи.


Таким образом, различные формы дисгенезии гонад обусловлены мозаичным кариотипом и отличаются особенностями клинической картины. Но для всех форм данной патологии имеются общие диагностические критерии: первичная аменорея; отсутствие или резкое недоразвитие вторичных половых признаков, генитальный инфантилизм; УЗИпризнаки дисгенетичных гонад; высокий уровень гонадотропинов, особенно ФСГ, соответствующий постменопаузальному возрасту; кариотип с аномальным набором половых хромосом, отсутствие или значительное снижение полового хроматина; отрицательная проба с гестагенами, но положительная с эстрогенами и гестагенами.


ЛЕЧЕНИЕ ЦЕЛИ ЛЕЧЕНИЯ Предотвращение малигнизации дисгенетичных гонад, находящихся в брюшной полости. Стимуляция пубертатного ростового скачка у больных с задержкой роста. Восполнение дефицита женских половых гормонов. Стимуляция и поддержание развития вторичных половых признаков для формирования женской фигуры. Активизация процессов остеосинтеза. Предупреждение возможных острых и хронических психологических, личных и социальных проблем. Профилактика бесплодия и подготовка к деторождению путём ЭКО донорской яйцеклетки и ПЭ.

Терапия дисгенезии гонад зависит от наличия в кариотипе Y хромосомы. В связи с высоким риском малигнизации гонад при её наличии необходимо их оперативное удаление эндоскопическим доступом в возрасте до 20 лет. При отсутствии в кариотипе Y хромосомы или после оперативного удаления гонад проводят заместительную гормональную терапию, которая направлена на: феминизацию фигуры, развитие полового оволосения, молочных желёз, матки; подавление уровня гонадотропинов; развитие циклических изменений в эндометрии с менструальной реакцией; профилактику эстроген дефицитных состояний (остеопороза, метаболических нарушений, сердечно сосудистых заболеваний); социальную адаптацию; улучшение качества жизни.